Frontier Sciences: KAMATANI Yoichiro

Over 99% of the human genome base sequence between individuals is identical, meaning that less than 1% is different. These differences are variants. Most human diseases show familial aggregation. This is often because a specific genetic sequence tends to develop a particular disease.
To identify the genes responsible for rare hereditary diseases, including congenital diseases, it is essential to analyze genome data from families in which such diseases are common. On the contrary, statistical analysis of massive genome samples is required to identify susceptibility genes (i.e., responsible genes) for common diseases. To date, we have identified numerous, at least tens of, susceptibility variants per disease by conducting statistical genetics analyses, including genome-wide association studies (GWAS), using genome data of several hundred thousand Japanese individuals stored at BioBank Japan (BBJ).
One of the reasons to identify disease susceptibility variants is that we can predict disease onset by accumulated genetic score (i.e., polygenic score). At present, Europe and the United States conduct larger-scale GWASs than in Japan and other East Asian countries. However, BBJ has reported that the accuracy of disease onset prediction for Japanese using GWAS results of European ancestry is unreliable and vice versa.
Although only approximately 1.5% of the human genome is protein-coding regions, most of the variants responsible for rare hereditary diseases are found there. Consequently, we can identify which genes are the determinants of disease. Responsible variants can result in incompletely synthesized proteins, damage or rebuilding of exon-intron boundaries, or altered biological activity due to changes to the tertiary structures of proteins caused by amino acid replacement. Therefore, only analysis of gene sequences may allow us to estimate how these variants may affect biological pathways in vivo.
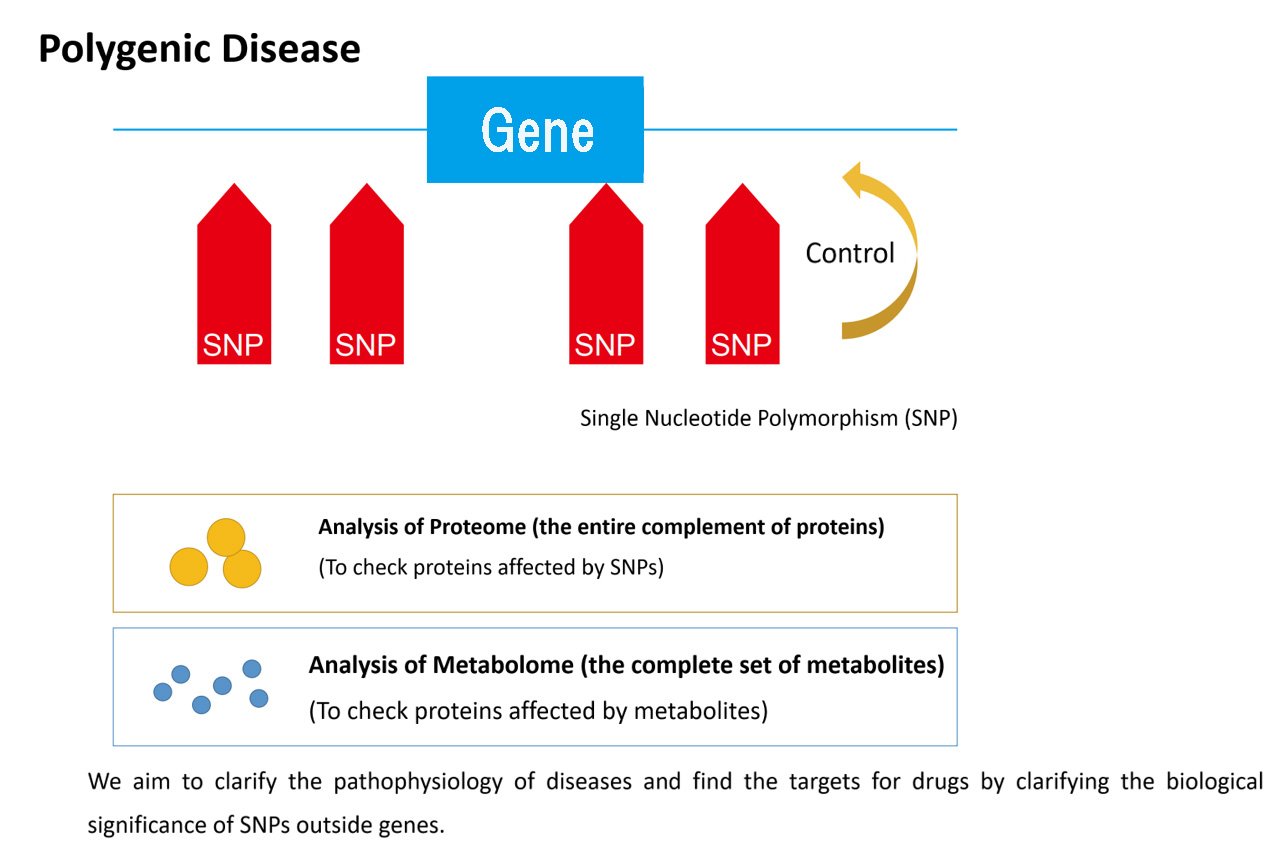
On the contrary, susceptibility variants for common diseases can also be found in the region outside of the gene. Additionally, improvements in epigenomic research have provided new information regarding noncoding regions. Consequently, it has become evident that most of the 98.5% of the genome that consists of noncoding regions still has some significant functionality. Moreover, susceptibility variants for common diseases can also be found in gene regulatory regions. This suggests that diversification of sequences in regulatory regions can affect gene expression and quantity of protein synthesis, resulting in increased risk of disease.
Currently, we aim to clarify the biological significance of susceptibility variants for common diseases by analyzing proteome data (i.e., comprehensive datasets related to protein) and metabolome data (i.e., comprehensive data from small molecules metabolite) obtained from blood samples collected by the BBJ. We then integrate these datasets with genome data to determine the extent to which genetic variants may impact protein levels or metabolic pathways.
Based on this research, we expect to comprehensively understand the genetic mechanisms responsible for common diseases when they are expressed as variations in DNA sequences. We then aim to clarify the morbid state and to identify possible pharmaceutical targets. Finally, we also strive to enable more efficient prediction of the development of common diseases despite challenges related to the demographic skew of existing datasets.

Communications with lab members are essential. (Left: Associate Professor Masaru Koido)
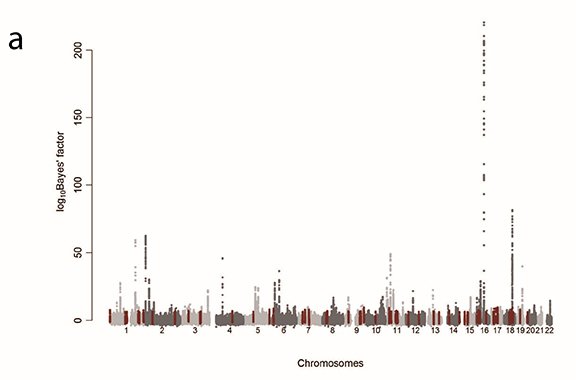
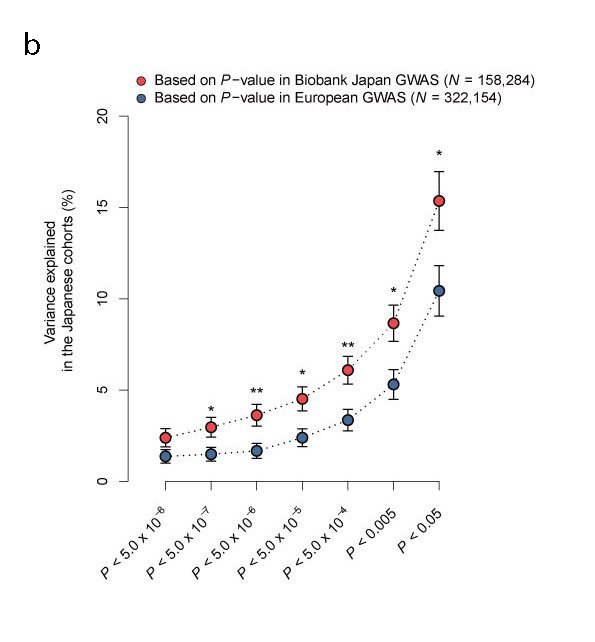
GWAS found numerous, at least tens of, susceptibility variants per disease or character. However, many of these were located outside of genes (a). We also note that the accuracy of polygenic scores may be undependable across different populations (b). Therefore, we investigate proteins, which are the results of heritable diversity in noncoding regions and in metabolites whose abundance is affected by proteins. In doing so, we aim to clarify the biological mechanism of heritable diseases, identify drug targets, and develop a race-agnostic disease prediction system.

KAMATANI Yoichiro
Professor
Laboratory of Complex Trait Genomics, Department of Computational Biology and Medical Sciences
vol.41
- Cover
- REDEFINE THE VALUE OF WATER FOR BETTER SOCIAL SYSTEMS
- Realizing Advanced Nuclear Fusion and Creating Antimatter Plasma: Learning From Natural Phenomena
- Clarifying the Biological Significance of Susceptibility Variants for Common Diseases
- Formulating Visually Perceived Environments: Impressions of Interior and Light Environment
- GSFS FRONTRUNNERS: Interview with an entrepreneur
- Voices from International Students
- ON CAMPUS x OFF CAMPUS
- EVENT & TOPICS
- INFORMATION
- Relay Essay