体細胞における反復配列間の組換えを解析 -ヒトゲノムの持つ新たな複雑性を発見-
- ヘッドライン
- 記者発表
理化学研究所
東京大学
産業技術総合研究所
発表概要
理化学研究所(理研)生命医科学研究センタートランスクリプトーム研究チームのジョバンニ・パスカレッラ研究員、ピエロ・カルニンチチームリーダー、東京大学大学院新領域創成科学研究科メディカル情報生命専攻生命システム観測分野の鈴木穣教授、産業技術総合研究所人工知能研究センターのフリス・マーティン上級主任研究員らの国際共同研究グループは、健常人や疾患を持つ患者の体細胞のゲノム上に存在する「反復配列[1]」間で起こる組換えを網羅的に解析し、ヒトゲノムの持つ新たな特徴を発見しました。
ヒトゲノムの50%以上は、特定の塩基配列が繰り返し出現する反復配列から構成されています。遺伝性疾患やがんの患者では類似性が高い反復配列間の組換えにより生じた変異がしばしば検出されることが報告されていました。
今回、国際共同研究グループは、体細胞ゲノムの反復配列間の組換えを系統的に見つけるための手法を独自に開発し、健常人の肝臓、腎臓、脳において、反復配列間の組換えによる変異が組織特異的に多く見られることを発見しました。また、ヒト細胞の分化が組換えに影響すること、アルツハイマー病やパーキンソン病などの神経変性疾患に反復配列の組換えによる変異が関わっていることを明らかにしました。これは、ゲノム上の反復配列間の組換えが遺伝性疾患やがんの患者のみで限定的に起こる現象ではなく、健常人や神経性疾患などでも起こる普遍的な現象であることを示しています。
本研究成果は、反復配列の組織特異的組換えがヒトゲノム構造および疾患へ与える影響についての体系的な理解につながると期待できます。
この研究は、科学雑誌『Cell』オンライン版(7月25付:日本時間7月26日)に掲載されました。
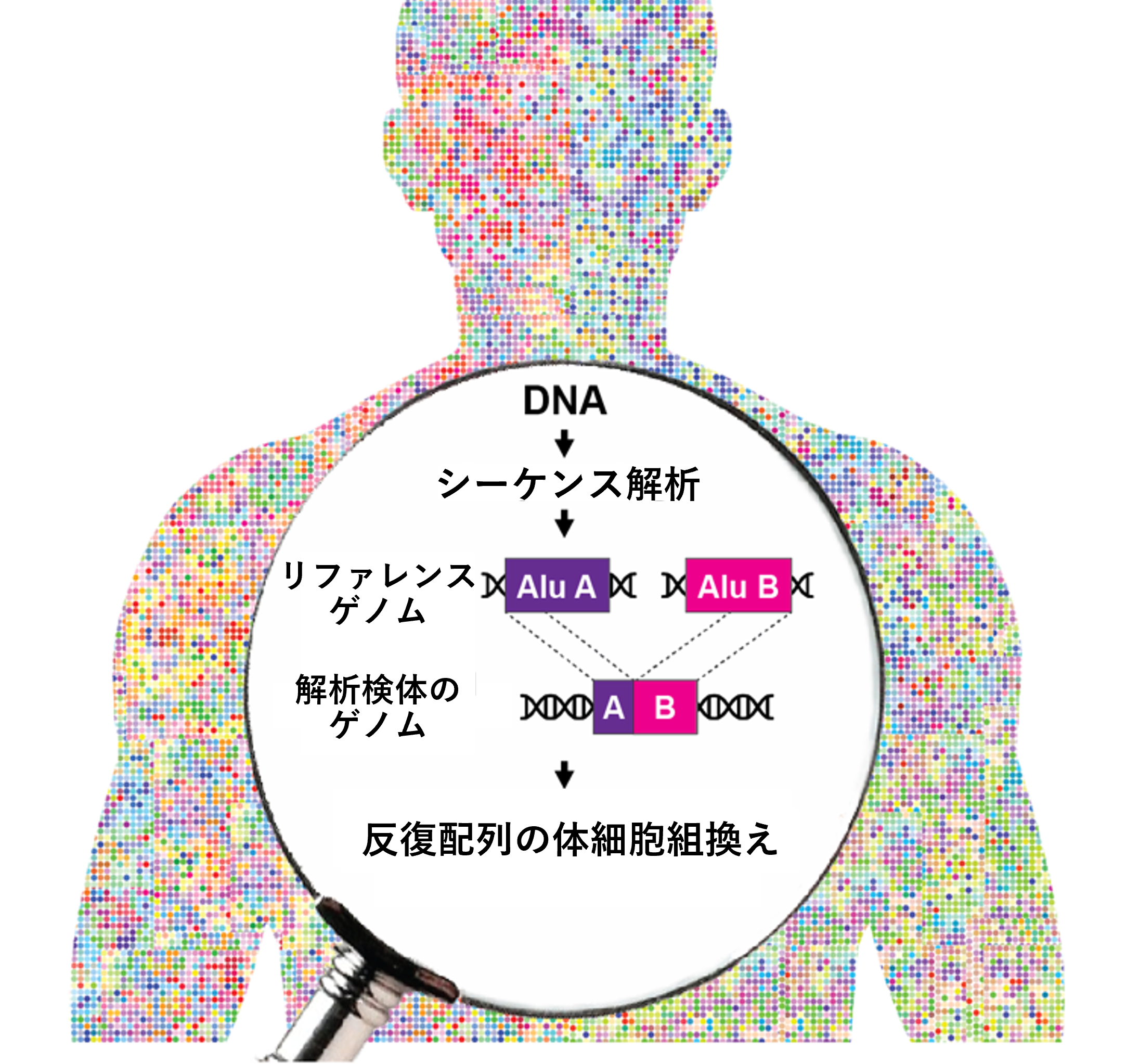
体細胞ゲノムにおける反復配列間の組換えによる変異を発見
研究内容
背景
20年以上前にヒトゲノムのドラフト配列が最初に解読された際、特定の塩基配列を何百万回も繰り返し出現する「反復配列」がゲノムの50%以上を構成していることが明らかになりました。
ヒトゲノム上の反復配列は、長さ300塩基対ほどの配列から構成される「Aluファミリー[2]」やおよそ7,000塩基対の配列から構成される「L1ファミリー[2]」などに分類されます。近年の研究により、これらの反復配列が近くの遺伝子の発現制御やゲノムの立体構造の形成など、生体内の重要なメカニズムに関わっていることが分かってきました。一方で、反復配列同士は類似性が高いため、反復配列間で組換えによる変異を引き起こします。実際、がんや遺伝性疾患の患者のゲノムでは、AluやL1反復配列の組換えによる遺伝子変異が見つかっています。しかし、このような特定のタイプのDNA変異に関して、さまざまな生体組織を用いた体系的な調査はこれまで行われていませんでした。
研究手法と成果
国際共同研究グループは、「がんや遺伝性疾患において報告されているAluおよびL1反復配列の組換えは氷山の一角にすぎず、実際にはもっと普遍的に起こる現象ではないか」という仮説を立てました。
はじめに、この仮説を実証するために、DNAサンプルから特定のゲノム配列を濃縮する実験手法「Capture-seq法[3]」を用いて、ヒトゲノム全体にわたる反復配列間の組換えを解析しました。この際、従来のCapture-seq法における反復配列に結合するRNA分子(プローブ)を独自設計し、短時間で効率的にDNA断片を濃縮できるように改良を加えました。これにより、組換えを包括的に検出することが可能になりました。
次に、この手法を用いて、東京都健康長寿医療センターの高齢者ブレインバンクから提供された10人の死後ドナーのDNA解析を行いました。それぞれのドナーについて、腎臓、肝臓、および3種類の脳部位(前頭皮質、頭頂皮質、側頭皮質)の組織からDNAを濃縮しました(図1上)。これらの検体からCapture-seq法により反復配列のDNA断片を抽出し、イルミナ次世代シーケンサー[5]を用いて、塩基配列を解析しました(図1右下)。また、同一組織のDNAを、キャプチャー法による濃縮やPCR増幅処理を行わずに、長鎖DNAの解析が可能なナノポアシーケンサー[6]により、全ゲノム配列解析しました(図1左下)。独立した2種類の解析手法を用いることで、解析結果の有意性が相互に検証可能となります。
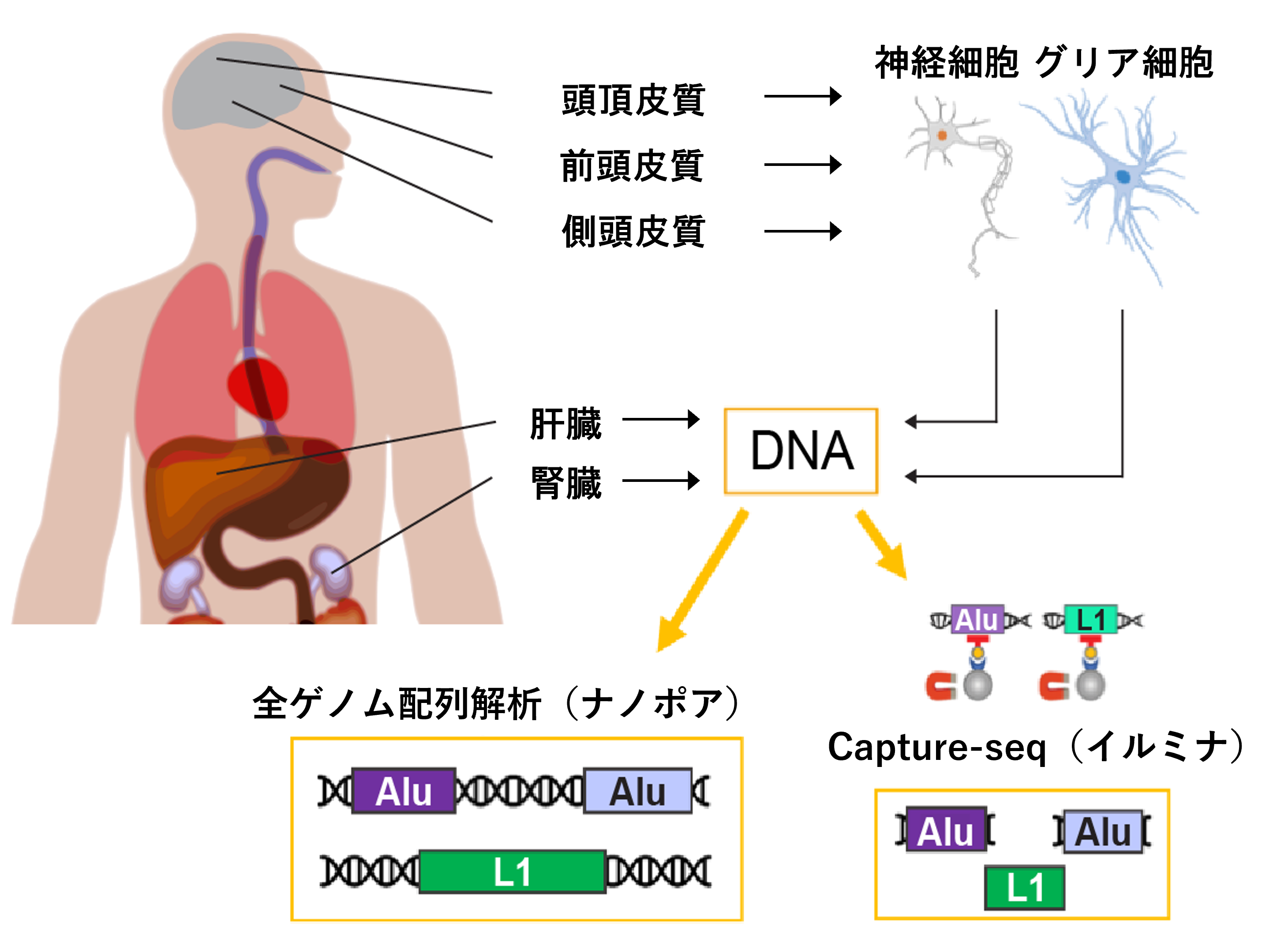
図1 本研究における実験的アプローチ
AluおよびL1反復配列の体細胞組換えを解析するために、腎臓、肝臓、3種類の脳部位(神経細胞とグリア細胞[4]別)のDNAについて、Capture-seq法により濃縮された反復配列DNA断片のイルミナ次世代シーケンサーによる配列解析およびナノポアシーケンサーによる全ゲノム配列解析を行った。
{kind=link}
さらに、シーケンス解析後の塩基配列データからAluおよびL1反復配列を抽出するための計算ツールとして、バイオインフォマティクスパイプライン[7]「TE-reX」を開発しました。このパイプラインでは、検体から得られたシーケンス配列データを、ヒトのリファレンスゲノム[8]配列データと比較し、完全に一致しないリードを抽出することで、組換えが起こった部位を同定します(図2)。
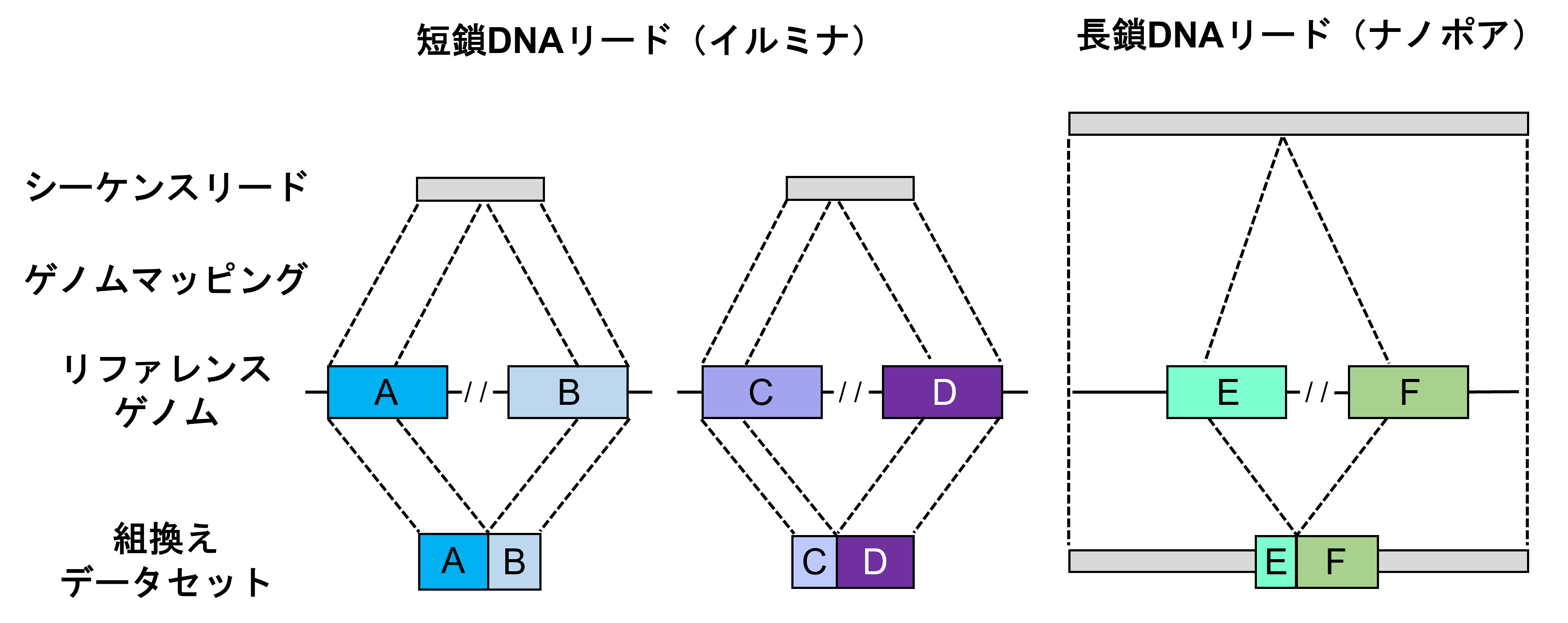
図2 TE-reXによる反復配列間の組換えの検出スキーム
TE-reX、リファレンスゲノム上の離れた位置に別々にマップされた検体由来のシーケンス配列を識別し、解析対象サンプルの組換え部位を抽出する。TE-reXを用い、図1で示したCapture-seq法により濃縮したDNA断片、および全ゲノム配列における組換えを検出した。
この方法で、ヒト検体から得られたシーケンスデータから、AluおよびL1反復配列の組換えにより生じた数百万カ所の変異を発見しました。さらに、それぞれの体細胞に特異的な組換えについて解析し、組換えの特徴を明らかにしました。例えば、腎臓および肝臓では1細胞当たりの組換え数が比較的多く(図3A)、脳組織では同一の染色体内での組換えが多く見られました。また、脳組織においては、ゲノム上の隣接した領域で、AluおよびL1反復配列が関与する組換えが多く見られました(図3B)。
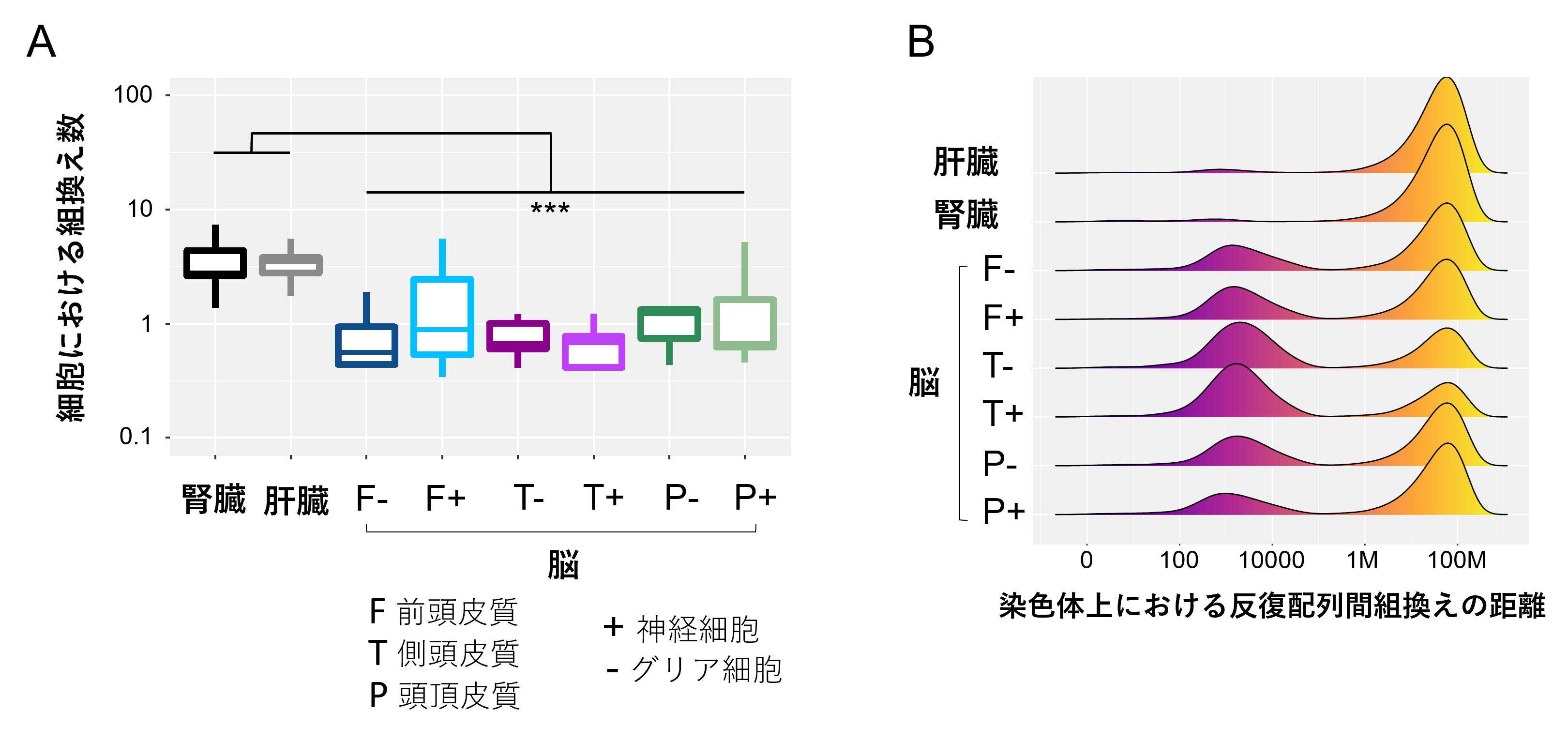
図3 体細胞における組織特異的な組換えの特徴
A:脳組織に比べて、腎臓および肝臓では1細胞当たりの組換え数が多かった。
B:縦軸は、横軸で示した距離だけ離れた反復配列間由来の組換えイベントの割合を組織ごとに示す。脳組織では、同一ゲノム上の近接する領域上での反復配列間の組換え頻度が高かった。
ここで、組換えの特徴が組織によって異なることから、「発生初期の生物学的なプロセスが反復配列の組換えに影響しているのではないか」と仮説を立てました。
この仮説を実証するため、iPS細胞(人工多能性幹細胞)[9]およびiPS細胞から分化誘導した神経細胞について、AluおよびL1反復配列の組換えをCapture-seq法とTE-reXを用いて解析しました。その結果、iPS細胞から分化誘導した神経細胞は、iPS細胞と比べ、不活性なクロマチンコンパートメント[10]における反復配列間の組換えの増加や近接した反復配列間の組換えの増加などの特徴を持つことが分かりました。これは、ヒト細胞の分化が体細胞における組換えに影響することを示しています。
また、神経変性疾患であるアルツハイマー病およびパーキンソン病の患者から提供された検体の組換えを解析したところ、疾患検体に特異的ないくつかの特徴を発見しました。アルツハイマー病のドナーでは、側頭皮質で組換えが増加することが分かりました。パーキンソン病のドナーでは、パーキンソン病に強く関連する遺伝子において、DNA欠失を引き起こす組換えが頻繁に見られました。これらの発見により、神経変性疾患が組換えの特異的な変化に影響を与える可能性が示唆されました。
今後の期待
本研究により、体細胞におけるAluあるいはL1反復配列の組換えが、健常人のヒトゲノムで見られる普遍的な特徴であることが示されました。これらの体細胞における組換えは、ヒトゲノムの構造や機能に影響を与える可能性があります。
分化過程で組織によって組換えの特徴が著しく異なるという発見は、体細胞におけるAluおよびL1反復配列間の組換えが生物の正常な発生に必要である可能性を示しています。さらに、アルツハイマー病およびパーキンソン病において組換えのプロファイルが変化することは、組換えの変化が脳疾患に影響を与えることを示唆しています。今後、さまざまな疾患における反復要素間の組織特異的組換えの特徴を評価することにより、疾患の原因を解明し、新たな治療法が開発されるものと期待できます。
発表雑誌
雑誌名:Cell
タイトル:Recombination of repeat elements generates somatic complexity in human genomes
著者:Giovanni Pascarella, Chung Chau Hon, Kosuke Hashimoto, Annika Busch, Joachim Luginbühl, Callum Parr, Wing Hin Yip, Kazumi Abe, Anton Kratz, Alessandro Bonetti, Federico Agostini, Jessica Severin, Shigeo Murayama, Yutaka Suzuki, Stefano Gustincich, Martin Frith and Piero Carninci.
DOI:10.1016/j.cell.2022.06.032
用語解説
[1] 反復配列
ゲノム上で特定の塩基配列が繰り返し出現する配列のことを指す。反復配列には、同じ配列が同じ向きに隣り合って存在するタイプ(縦列反復配列)と、隣り合わずに存在するタイプ(散在反復配列)などがある。
[2] Aluファミリー、L1ファミリー
Aluファミリーは長さ300塩基対ほどの短鎖散在反復配列で、ヒトゲノムの約10%を占める。L1ファミリーは7,000塩基対ほどの長鎖散在反復配列で、ヒトゲノムの約17%を占める。
[3] Capture-seq法
特定のゲノム領域を含むDNA断片を濃縮し、シーケンス解析を行う手法。目的とする領域と相補的な塩基配列を持つプローブを用いて、特定DNA断片をハイブリダイゼーション法により捕捉する。本研究では、反復配列に対するプローブを独自設計することで、従来法より濃縮にかかる時間を大幅に短縮し、高い効率でより希少な変異を検出できるように改良を加えた。
[4] グリア細胞
神経膠(しんけいこう)細胞とも呼ばれる。脳内の神経細胞を除いた多くの細胞の総称で、アストロサイト、オリゴデンドロサイト、ミクログリアなどがある。
[5] イルミナ次世代シーケンサー
Illumina社が開発した次世代シーケンサー。蛍光ヌクレオチドを使ったSequence by synthesis(シーケンス・バイ・シンセシス)に代表される手法を活用して、DNAの塩基配列を読み出す装置であり、サンガー法と比較すると大幅に高いスループットを達成する。
[6] ナノポアシーケンサー
Oxford Nanopore Technologies社が開発したシーケンサー。ナノスケールの穴を持つタンパク質をDNA分子が通過する際の電気信号から塩基配列を読み取る方法。現在次世代シーケンサーとして広く用いられているIllumina社の読み取り方法と比較すると、DNA分子の断片化を経ることなく長鎖のまま解析できることが特長である。
[7] バイオインフォマティクスパイプライン
バイオインインフォマティクスは生物が持っているさまざまな情報を計算機で解析する分野。計算処理のステップを分割してそれぞれが独立した処理を行ったものを接続するパイプライン手法を、同分野に適用することで、解析の最適化・高速化が図れる。
[8] リファレンスゲノム
ある生物種のゲノム配列として、標準ゲノム配列として公開されているもの。リファレンスにリードデータをアライメントすることにより、標準リファレンス配列と異なるDNA多型が検出される。本研究では、ゲノムリファレンスコンソーシアムが公開している総塩基数約3GbのヒトゲノムリファレンスGRCh38を用いた。
[9] iPS細胞(人工多能性幹細胞)
脊椎動物の初期胚などが持つ、全ての種類の体細胞へ分化する能力を多能性という。多能性を持ち、試験管内で培養して無限に増やすことができる細胞を多能性幹細胞という。iPS細胞は、成人の血液や皮膚の細胞にOct3、Sox2、Klf4遺伝子などを導入して人工的に作製した多能性を持つ幹細胞である。
[10] クロマチンコンパートメント
DNAとタンパク質の複合体であるクロマチンは高度に折り畳まれ、クロマチンコンパートメントと呼ばれる核内の領域に配置される。細胞核内ではAおよびBコンパートメントが領域を形成し、前者では遺伝子の転写が活性化され、後者では抑制されている。核内のコンパートメントは細胞分化や細胞周期によって変化することが分かっている。
国際共同研究グループ
理化学研究所 生命医科学研究センター
トランスクリプトーム研究チーム
チームリーダー ピエロ・カルニンチ (Piero Carninci)
(ヒューマン・テクノポール チームリーダー)
研究員 ジョバンニ・パスカレッラ(Giovanni Pascarella)
研究員(研究当時) 橋本 浩介 (ハシモト・コウスケ)
(現 大阪大学 蛋白質研究所 計算生物学研究室 准教授)
客員技師 ウィン ヒン・イップ(Wing Hin Yip)
客員研究員 アレッサンドロ・ボネッティ(Alessandro Bonetti)
テクニカルスタッフ(研究当時) アニカ・ブッシュ (Annika Busch)
ゲノム情報解析チーム
チームリーダー ヂョン チョウ・ホン(Chung Chau Hon)
遺伝子制御回路研究チーム
研究員 カラム・パール (Callum Parr)
研究員(研究当時) ジョアヒム・ルーゲッンブル(Joachim Luginbühl)
応用計算ゲノミクス研究チーム
技師 ジェシカ・セヴェリン(Jessica Severin)
東京大学 大学院新領域創成科学研究科 メディカル情報生命専攻 生命システム観測分野
教授 鈴木 穣 (スズキ・ユタカ)
産業技術総合研究所 情報・人間工学領域
上級主任研究員 フリス・マーティン (Frith Martin)
(東京大学 大学院新領域創成科学研究科 メディカル情報生命専攻 大規模バイオ情報解析分野 教授)
カロリンスカ研究所
研究員 フェデリコ・アゴスティーニ(Federico Agostini)
カリフォルニア大学 サンディエゴ校
アシスタントリサーチャー アントン・クラウツ (Anton Kratz)
イタリアン・インスティチュート・オブ・テクノロジー(IIT)
アソシエイトディレクター ステファノ・ガステンチッチ(Stefano Gustincich)
東京都健康長寿医療センター研究所
研究部長 村山 繁雄 (ムラヤマ・シゲオ)
研究支援
本研究は,日本医療研究開発機構(AMED)脳科学研究戦略推進プログラムにおける研究開発課題「老化・認知症拠点の構築」(研究開発代表者:村山繁雄)の支援を受けて行われました。また、日本学術振興会(JSPS)科学研究費助成事業新学術領域研究(研究領域提案型)『学術研究支援基盤形成』「コホート・生体試料支援プラットフォーム(CoBiA)」(中核機関:東京大学医科学研究所)の試料提供・解析支援を受けました。