大腸がんの「ゲノム医療」に貢献 - 日本人での原因遺伝子・発症リスク・臨床的特徴の大規模解析 -
- ヘッドライン
- 記者発表
理化学研究所
東京大学医科学研究所
東京大学大学院新領域創成科学研究科
埼玉県立がんセンター
発表概要
理化学研究所(理研)生命医科学研究センターがんゲノム研究チームの藤田征志上級研究員(研究当時)、中川英刀チームリーダー、基盤技術開発研究チームの桃沢幸秀チームリーダー、東京大学医科学研究所の村上善則教授、東京大学大学院新領域創成科学研究科松田浩一教授、埼玉県立がんセンターがんゲノム医療センターの赤木究センター長らの共同研究グループ※は、世界最大規模となる大腸がん患者を含む日本人集団36,000人以上のDNAを解析し、日本人遺伝性大腸がんの原因遺伝子・発症リスク・臨床的特徴を明らかにしました。
本研究成果は、日本人の遺伝性大腸がんの診断および患者一人一人に合った治療を行う「ゲノム医療」に貢献すると期待できます。
大腸がんは日本人の罹患数が部位別に男性で3番目、女性では乳がんに続いて2番目に多いがんです。乳がんや前立腺がんなどと同様に、大腸がん患者の数%は一つの「病的バリアント[1]」が発症原因であると考えられています。しかし、日本人大腸がん患者の病的バリアントについての大規模なデータは少なく、日本人集団での病的バリアントの影響は明確ではありませんでした。
今回、共同研究グループは、米国国立包括がんネットワーク(NCCN)ガイドラインで検査が推奨されている12個の遺伝子を含む計27個の遺伝性腫瘍関連遺伝子を対象に、独自に開発したゲノム解析手法を用いて、バイオバンク・ジャパン[2]により収集された大腸がん患者12,503人および対照群23,705人の血液のDNAを解析しました。その結果、397個の病的バリアントを同定し、大腸がん発症に関連するとされるDNAミスマッチ修復遺伝子[3]および遺伝性乳がん、卵巣がんの原因遺伝子であるBRIP1やBRCA1/2を含む8個の遺伝子が日本人集団における大腸がん発症に関連することや、病的バリアント保有者に見られる発症リスク、臨床的特徴を明らかにしました。今後、本研究で同定したバリアントデータは国内外の公的データベースに登録、活用される予定です。
本研究は、科学雑誌『Clinical Gastroenterology and Hepatology』(8月24日付)に掲載されました。
大腸がんのゲノム医療
発表内容
背景
大腸がんは欧米や日本で罹患数の多いがんの一つであり、日本人では罹患数が部位別に男性で3番目、女性では乳がんに続いて2番目に多いがんです注1)。大腸がん患者の約20%は、親族に大腸がんや他のがん患者を持つといわれ、大腸がんの遺伝性が強いことが示唆されています。ゲノム配列には、個人間の1塩基の違いである「遺伝的バリアント[1]」がありますが、それらの中でも特定の遺伝子上に存在し疾患発症リスクを大きく上げる「病的バリアント」は、遺伝性大腸がんの発症原因として挙げられます。
ゲノム上の病的バリアントの有無を調べる遺伝学的検査は、次世代シーケンサー[4]の進歩により、1遺伝子を検査する方法から発展し、現在ではさまざまな遺伝性疾患に関係する複数の遺伝子を同時に検査できるパネル検査が生まれ、費用対効果が高くなりました。遺伝学的検査は、世界最大規模の米国国立包括がんネットワーク(NCCN)注2)のガイドラインでも推奨されており、その対象となる遺伝子も既に公開されていますが、それらの遺伝子は主に欧米人集団の研究結果をもとに設定されています。各遺伝子の疾患発症への寄与の度合いは人種によって異なることが分かっており、欧米人集団の病的バリアントのデータをもとに作成されたガイドラインが日本人集団にも適用できるかどうかは、日本人集団の大規模なデータを解析して検証する必要がありました。
また、遺伝学的検査を受ける患者には経済的・心理的な負担が伴うだけでなく、その負担は患者の近親者にも及びます。そのため、疾患発症の原因を特定するための検査が必要な患者をあらかじめ絞り込むことは、患者や近親者の負担を軽減することにつながります。また、病的バリアント保有者に見られる臨床的な特徴が分かれば、遺伝学的検査前の臨床情報から病的バリアント保有の可能性のある患者を絞り込むことができます。しかし、絞り込む基準を設けるために必要な日本人集団の病的バリアントと臨床情報の関係についてのデータや、解析方法を評価したデータはほとんど蓄積されていませんでした。
そこで、共同研究グループは、日本人集団の大腸がんについて大規模なサンプルを用いた解析により、原因遺伝子や病的バリアントを同定し、遺伝性大腸がんの頻度を調べることで、日本人患者やその家族に向けたゲノム医療につながるデータを蓄積することを目指しました。さらに、今回の解析で検出された病的バリアントのデータおよびバイオバンク・ジャパンから提供された臨床データを用いて、日本人集団における病的バリアント保有者を絞り込むための解析方法の評価を行いました。
注1)国立がんセンター がん情報サービス がんの統計 2022
https://ganjoho.jp/public/qa_links/report/statistics/index.html
注2)https://www.nccn.org/professionals/physician_gls/default.aspx
研究手法と成果
共同研究グループはまず、バイオバンク・ジャパンにより収集された大腸がん患者12,503人および対照群23,705人(合計36,208人)の血液DNAを、理研が独自に開発したゲノム解析手法注3)を用いて解析しました。解析対象の遺伝子は、NCCNガイドラインで検査が推奨されている12遺伝子および研究開始当時に25遺伝子遺伝性がんパネル注4)で検査対象となっていた25遺伝子から選抜した15遺伝子の計27個の遺伝性腫瘍関連遺伝子としました。解析の結果、4,804個の遺伝的バリアントを同定しました。さらに、これらの遺伝的バリアント一つ一つを米国臨床遺伝・ゲノム学会(ACMG)注5)および分子病理学会(AMP)注6)が作成したガイドラインや、国際的データベースであるClinVar注7)の情報に基づいて、病的バリアントか否か評価しました。その結果、397個の遺伝的バリアントが病的バリアントであると判定され、そのうち199個(50.1%)はClinVarに未登録の新しい病的バリアントであり、日本人に特異的な病的バリアントである可能性があります。
次に、同定した397個の病的バリアントを、患者群・対照群それぞれがどのくらいの割合で保有しているか調べたところ、患者群の3.3%、対照群の1.6%が保有しており、病的バリアントを持つことで大腸がんへのなりやすさ(発症リスク)が約2.2倍高まることが分かりました(表1)。遺伝子ごとの解析では、8遺伝子(MSH2、MLH1、MSH6、APC、BRIP1、BRCA1、BRCA2、TP53)が日本人集団の大腸がん発症に寄与していることが統計学的に明らかになりました(表1)。なかでも、MSH2(p=6.0x10-14)、APC(p=1.7x10-11)、MSH6(p=2.1x10-8)、MLH1(p=1.6x10-10)の4遺伝子が大きく寄与しており(表1)、そのうち3個の遺伝子(MSH2、MSH6、MLH1)はDNAミスマッチ修復遺伝子でした。また、BRCA1(p=0.0034)、BRCA2(p=0.0041)の2遺伝子は、日本人集団における乳がん、前立腺がん、膵臓がんとの関連が既に明らかになっており、ヨーロッパやアジアの研究でも関連が示唆されています。一方で、BRIP1遺伝子(p=0.0034)は、欧米では乳がんや卵巣がんと関連があるものの大腸がんとの関連は報告されておらず、本研究で初めて関連が示されました。
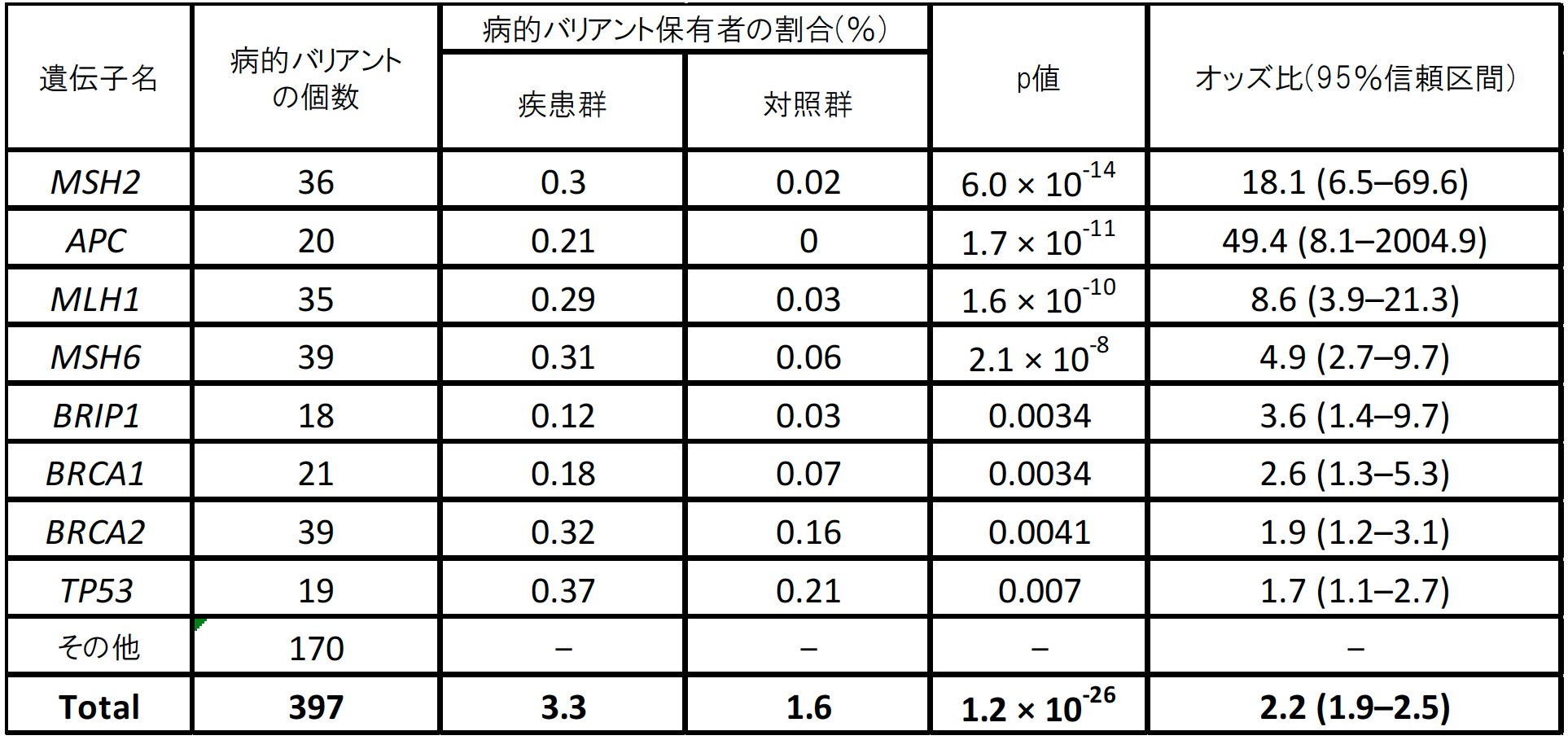
表1 本研究で明らかになった大腸がん原因遺伝子別の病的バリアント保有者と発症リスク
病的バリアント保有者の割合は、患者群で3.3%、対照群で1.6%であり、オッズ比は2.2倍であった。オッズ比は、ある事象の起こりやすさについて二つの群で比較したときの違いを示す統計学的尺度の一つで、病的バリアントを持つことで大腸がんへのなりやすさ(発症リスク)が2.2倍高まることを示す。この表におけるp値は、偶然にそのようなことが起こる確率のことで、統計学的有意差を示す指標である。p値が低いほど偶然では起こり得ないことを示す。
次に、大腸への発がんリスクを高めるリンチ症候群[5]の主な原因となるDNAミスマッチ修復遺伝子注8)上の「コピー数多型(CNV) [6]」を特定するため、以前の研究で用いられた4種類の高密度DNAマイクロアレイ[7]データを使用し、CNVを検出するソフトウェアを用いた再解析および定量PCR[8]を行いました。その結果、3遺伝子(EPCAM-MSH2、MLH1、APC)上に30個のCNVが検出され、そのうち23個が欠失[9]、7個が重複[10]でした。さらに、30個のうち23個をMLPA法[11]で再確認したところ、18個が翻訳領域に影響していることが分かり、「病的CNV」であると評価しました。
また、病的バリアント保有者に臨床的な特徴があるかどうかを、大腸がんの病的バリアント保有患者416人と非保有患者12,087人を比較することで調べました。その結果、病的バリアント保有患者の数には、診断時の年齢が下がるにつれて有意な上昇が見られ(図1)、保有患者の大腸がん診断年齢は、非保有患者に比べて平均3.0歳若いことが分かりました。加えて、保有患者では、4種類のがん(大腸がん、乳がん、子宮内膜がん、卵巣がん)の家族歴や3種類のがん(胃がん、子宮内膜がん、卵巣がん)の既往歴を持つ人が、対照群よりも多いことが統計学的に明らかになりました。
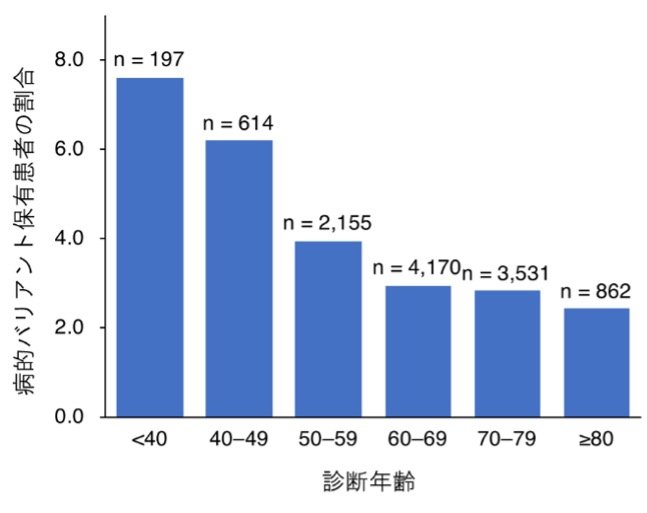
図1 大腸がんにおける診断年齢ごとの病的バリアント保有患者の割合
40歳未満で大腸がんと診断された患者の7.6%が病的バリアントを保有しており、最も割合が高かった。病的バリアント保有者の割合は、診断時の年齢が上がるにつれて有意な減少が見られた(P=2.3x10-7)。
さらに、遺伝学的検査の前に病的バリアント保有者を予測するために、2種類の解析モデルを検討しました。一つめは、実際の大腸がん患者に見られた家族歴や既往歴、診断年齢などの臨床的特徴を12項目設定し、その項目の合致数でスコアリングをする方法で、本研究で大腸がんへの関連が示された8遺伝子に病的バリアントを保有している患者と非保有患者で比較しました。その結果、病的バリアント保有患者のスコア(平均1.2)は、非保有患者のスコア(0.6)よりも有意に高くなりました。しかし、異なる閾値について感度・特異度をプロットしたROC曲線で解析モデルの予測精度を評価したところ、予測精度を表すAUCは0.6355であり(図2a)、実際に臨床現場で利用可能な精度ではありませんでした。二つめは、機械学習[12]技術を用いて8遺伝子の病的バリアント保有者の予測を試みました。臨床的特徴20項目を基にランダムフォレストなど6種類の解析方法を用いて予測精度を検討しました。その結果、方法によって予測精度にばらつきが見られたものの、一つめの方法と同程度もしくはそれ以下の精度であると評価されました(図2b)。
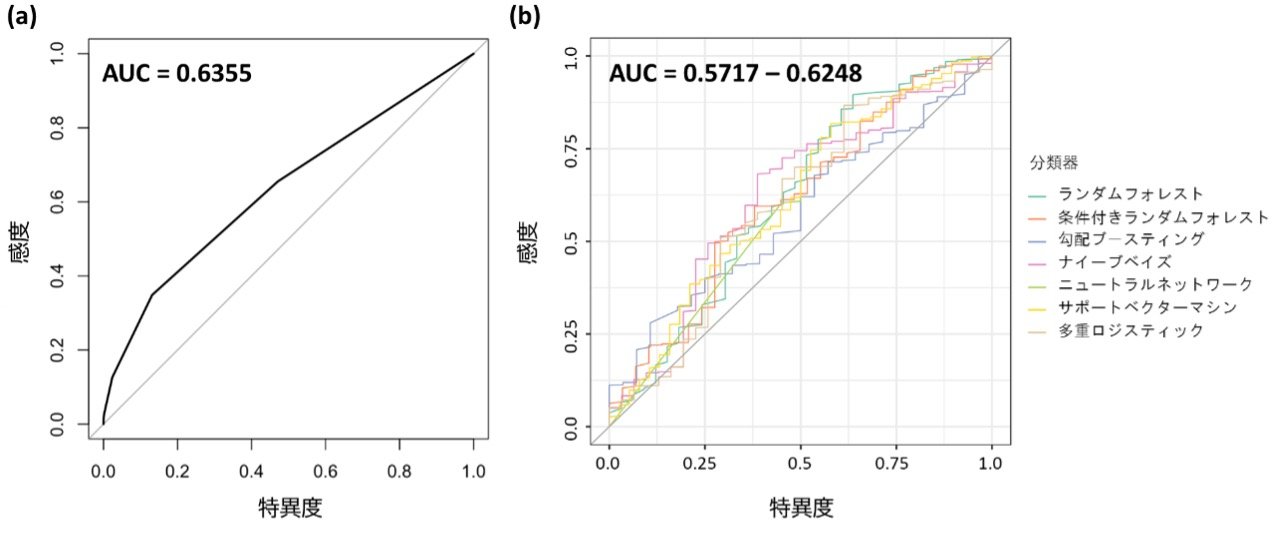
図2 ROC曲線を用いた解析モデルの精度検証
感度・特異度をそれぞれ縦軸・横軸にプロットしたROC曲線。予測モデルの精度を示し、曲線の下の面積が広い方が、より高精度に予測できることを示している。それを数値化したものがAUCスコアで、値が1に近いほど判別能が高いことを示す。
(a) 臨床的特徴12項目の合致数でスコアリングをする方法の結果。高い予測精度は得られなかった。
(b) 機械学習技術によって保有者の予測をする6種類の解析方法の結果。予測精度は(a)と同程度だった。
本研究で得られた各バリアントのサマリー情報は、日本の臨床ゲノム情報統合データベース(MGeND)のほか、海外のデータベースにも登録され、臨床現場における遺伝的バリアントの解釈について重要な情報として利用されます。
注3)2018年6月21日プレスリリース「炎症性腸疾患発症に関わる複雑な遺伝子発現制御機構」https://www.riken.jp/press/2018/20180621_1/index.html
注4)Myriad myRisk Hereditary Cancer panel
https://myriad.com/products-services/hereditary-cancers/myrisk-hereditary-cancer/
注5)American College of Medical Genetics
https://www.acmg.net/
注6)Association for Molecular Pathology
https://www.amp.org/
注7)https://www.ncbi.nlm.nih.gov/clinvar/
注8)Lynch HT, la Chapelle A de. Hereditary colorectal cancer.
N Engl J Med 2003;348:919-32.
今後の期待
本研究では、36,000人以上に及ぶ大腸がん患者群・対照群のDNAを用いた世界最大規模の解析を行い、日本人の大腸がん患者における病的バリアントや、遺伝性大腸がんと関連する8遺伝子および臨床的特徴を明らかにしました。これらの情報は、日本人集団へ向けた大腸がんの遺伝学的検査やガイドライン開発に貢献し、今後、日本人へ向けた遺伝性がんの検査体制や患者一人一人に合ったゲノム医療体制が構築されていくと期待できます。
研究支援
本研究は、日本医療研究開発機構(AMED)ゲノム創薬基盤推進研究事業「乳がん・大腸がん・膵がんに対する適切な薬剤投与を可能にする大規模データ基盤の構築(研究開発代表者:桃沢幸秀)」による支援を受けて行われました。
論文情報
タイトル:Population-based Screening for Hereditary Colorectal Cancer Variants in Japan
著者名:Masashi Fujita, Xiaoxi Liu, Yusuke Iwasaki, Chikashi Terao, Keijiro Mizukami, Eiryo Kawakami, Sadaaki Takata, Chihiro Inai, Tomomi Aoi, Misaki Mizukoshi, Kazuhiro Maejima, Makoto Hirata, Yoshinori Murakami, Yoichiro Kamatani, Michiaki Kubo, Kiwamu Akagi, Koichi Matsuda, Hidewaki Nakagawa, and Yukihide Momozawa
雑誌:Clinical Gastroenterology and Hepatology
用語解説
[1] 病的バリアント、遺伝的バリアント
ヒトのDNA配列は30億の塩基対からなるが、その配列の個人間の違いを遺伝的バリアントという。そのうち、疾患発症の原因となるものを病的バリアントと呼ぶ。
[2] バイオバンク・ジャパン
日本人集団27万人を対象とした、世界最大級の疾患バイオバンク。オーダーメイド医療の実現プログラムを通じて実施され、ゲノムDNAや血清サンプルを臨床情報と共に収集し、研究者へ分譲を行っている。2003年から東京大学医科学研究所内に設置されている。
[3] DNAミスマッチ修復遺伝子
細胞分裂の際のDNA複製時に塩基の不対合(ミスマッチ)を修復する機能を担うタンパク質をコードしている遺伝子のこと。
[4] 次世代シーケンサー
サンガー法を利用した蛍光キャピラリーシーケンサーである「第一世代シーケンサー」と対比させて使われる用語。機種によるが、1回に最大数兆塩基のDNAの配列を決定できる。
[5] リンチ症候群
がんの発症リスクを高める遺伝性疾患の一つで、主に大腸への発がんリスクを高めるとされている。ミスマッチ修復遺伝子の変異が原因であることが判明しており、米国国立包括がんネットワーク(NCCN)では、50歳未満の全ての大腸がん患者にはリンチ症候群の遺伝学的検査を受けることを推奨している。
[6] コピー数多型(CNV)
ゲノム配列の個人間の違いの一種で、ゲノム配列の一部の領域が失われる、もしくは増えて挿入されることによって、その部分のゲノム配列の存在数が増減すること。CNVはCopy Number Variationの略。
[7] 高密度DNAマイクロアレイ
1塩基の違いを検出する人工のゲノム配列が高密度に敷き詰められているチップのこと。マイクロアレイ上に検体のDNAを流すことによって、そのDNAに標的配列が含まれていることおよびその量を調べることができる。
[8] 定量PCR
ポリメラーゼ連鎖反応(PCR)により、標的の DNA 配列の増幅量を経時的に測定する方法。増幅量から、元の DNA のコピー数を定量できる。PCRはPolymerase Chain Reaction の略。
[9] 欠失
染色体または、DNAの塩基配列の一部が失われること。遺伝子機能に関わる部分に起こると、遺伝子機能の喪失を引き起こし、疾患に関わる場合がある。
[10] 重複
ゲノム配列の一部の領域が重複して挿入された形態。欠失に比べて数は少ないが、遺伝子機能に関わる部分に起こると、通常の遺伝子発現パターンより多く発現されることになるため、遺伝子機能の喪失を引き起こす欠失と同様、疾患との関わりが多く報告されている。
[11] MLPA法
標的のDNAの配列に結合する配列とPCR法で増幅されるプライマー配列が隣り合ったプローブを用いて、標的配列が存在するかを確認するための手法である。また蛍光強度の程度によって、存在する配列の重複や欠失などの標的配列のコピー数を判別できる。MLPAはMultiplex Ligation-dependent Probe Amplificationの略。
[12] 機械学習
コンピュータが大量のデータから規則性や判断基準を学習し、それに基づいて未知のデータを分類する技術。
関連研究室
新領域創成科学研究科 メディカル情報生命専攻 クリニカルシークエンス分野