非翻訳RNAの発現を予測するAIの開発-ゲノム解析からの疾患理解を促進-
- ヘッドライン
- 記者発表
理化学研究所
東京大学
産業技術総合研究所
株式会社ヒューマノーム研究所
発表概要
理化学研究所(理研)生命医科学研究センター ゲノム解析応用研究チームの小井土大 特別研究員(研究当時、現 客員研究員、東京大学 大学院新領域創成科学研究科 助教)、寺尾知可史 チームリーダー、ピエロ・カルニンチ 副センター長(研究当時)、東京大学 大学院新領域創成科学研究科の鎌谷洋一郎 教授、産業技術総合研究所(産総研)人工知能研究センターの瀬々潤 客員研究員(株式会社ヒューマノーム研究所代表取締役社長)らの共同研究グループは、300種以上の細胞・組織における非翻訳RNA[1]の発現をDNA配列パターンのみから予測するAI[2]を開発しました。
本研究成果は、創薬標的やバイオマーカーとなる非翻訳RNAの探索に貢献すると期待できます。
多因子疾患などに関連する多型[3]はゲノムの非翻訳領域に集積していますが、多型が非翻訳RNAの発現に与える影響の多くが未解明でした。
今回、共同研究グループは数十年前から行われてきた配列モチーフ[4]解析に立ち戻り、DNA配列パターンから非翻訳RNAの発現を予測するAIを開発し、「MENTR(メンター)」と名付けました。FANTOMコンソーシアム[5](FANTOM5)が2014年に公開した遺伝子発現データをMENTRで学習し、過去のゲノムワイド関連解析(GWAS)[6]の結果の再解釈を行いました。これにより、多因子疾患などのさまざまな形質に関連する非翻訳RNAを1万種以上カタログ化し、非常にまれな多型が非翻訳RNAを介して喘息などの疾患発症に影響する機序を明らかにしました。MENTRは公開され、世界中の研究者が利用可能です。
本研究は、科学雑誌『Nature Biomedical Engineering』オンライン版(11月21日付:日本時間11月22日)に掲載されました。
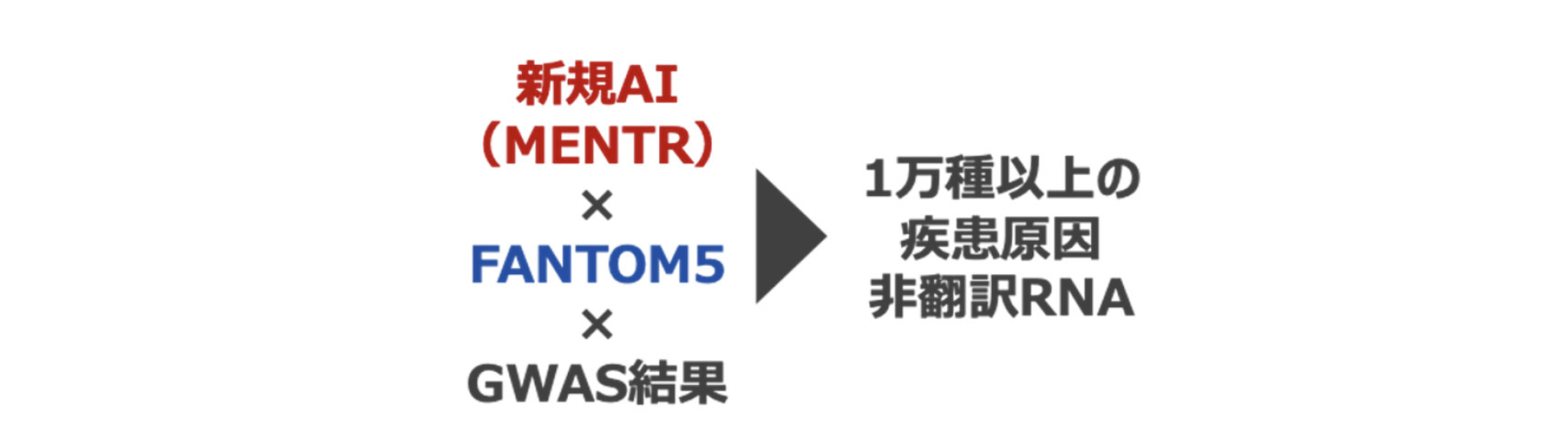
新しいAI(MENTR)を活用した非翻訳RNAの発現予測と疾患研究への応用
発表内容
研究の背景
世界中で数十万人以上の検体を用いたゲノムワイド関連解析(GWAS)が盛んに実施され、疾患のかかりやすさや、身長・体重・血液検査値などの複雑な形質に関連する遺伝多型が数多く同定されてきました。こうした遺伝的関連情報を用いることで、創薬の成功確率が上がるなどの証拠が蓄積されており、多型の生物学的機能の解明の重要性がますます高まっています。
これまでに、さまざまな複雑形質に関連する多型の多くはタンパク質に翻訳されないゲノム上の領域(非翻訳領域)に存在し、タンパク質に翻訳されるメッセンジャーRNA(mRNA)量の調節のみならず、非翻訳領域から転写されるさまざまなRNA(非翻訳RNA)量の調節を介して、形質の個人差に影響することが示されてきました。こうした非翻訳領域上の多型の役割を解明するには、多型とRNA量との関連を組織別に網羅的にまとめたデータベース(カタログ)が必要です。
しかし、mRNAに比べると、非翻訳RNAと多型との関連のカタログは不完全です。特に、mRNAの発現を遠隔から制御する機能領域(エンハンサー)が活性化して転写されるRNA(エンハンサーRNA:eRNA[7])は、非翻訳RNAの中でも特に発現量が低く、実験的定量が困難です。さらに、eRNAは同じDNA配列であっても細胞種ごとに非常に異なる発現パターンを示すため、さまざまな細胞での実験が必要といった課題がありました。そのため、eRNAがさまざまな細胞の機能に広く関わると考えられる一方で、eRNAと多型との関連を明らかにすることは極めて困難でした。
そこで共同研究グループは、分子生物学者が数十年前から経験的に見いだしてきた「配列モチーフ」という考え方をAI技術(特にディープラーニング[8])に適用して発展させる研究手法に着目しました。この手法では、ゲノム上の特定の連続したDNA配列パターン(モチーフ)と転写との関係性をAIで学習した後、コンピュータ上でモチーフが破壊された際の転写への影響を予測する「インシリコ変異導入法」を行います。本研究では、DNA配列と非翻訳RNAの発現パターンの関係を正しく学習させて新しいAIを開発し、多型による非翻訳RNAの細胞特異的な発現量変化を実験的変異導入法に匹敵する精度で予測することを目指しました。
研究手法と成果
共同研究グループは、一般に公開されているヒトの標準的なゲノム配列から取得した転写物周辺20万塩基のDNA配列を入力データとし、その配列の中央からRNAが発現する確率を出力するAIを設計し、FANTOM5が収集した300種以上のヒト初代細胞や組織のCAGEトランスクリプトームデータ[9]の発現パターンをAIに学習させました。
このAIは大きく2段階構成となっており、1段階目ではディープラーニングを用いて、DNA配列パターンから2,002種類のエピジェネティックな状態量[10](さまざまな転写因子[4]の結合など)を推定し、2段階目では勾配ブースティング決定木[11]によってエピジェネティックな状態量から細胞種ごとのRNA(mRNAと非翻訳RNA)の転写の有無を予測します。学習後のAIを用いると、任意のDNA配列(入力データ)から細胞・組織ごとのRNA発現確率が出力されます。興味深いことに、予測されたRNA発現確率は実際のRNA発現量にも相関します。さらに、DNA配列に変異を加えたときの出力値変化を観察するインシリコ変異導入法を行うと、変異による発現量変化を予測することができます。この非翻訳RNA予測に特化した手法を「MENTR(メンター:Mutation Effect prediction on ncRNA transcription)」と名付けました。
まず、MENTRがDNA配列から組織・細胞種ごとの非翻訳RNAの発現をどの程度正確に予測できるかを確かめるために、学習データと重複しない評価用データを用いて予測能(ROC曲線下面積[12])を評価しました。FANTOM5が収集・公開している347種類のさまざまな細胞・組織のCAGEトランスクリプトームデータにおいて、mRNAを0.87±0.02(ROC曲線下面積の平均と標準偏差、n=347)、長鎖ノンコーディングRNA(lncRNA)[1]を0.76±0.04、eRNAを0.69±0.05の精度で予測でき、特にlncRNAやeRNAの予測精度が従来法に比べて大幅に改善しました(図1)。
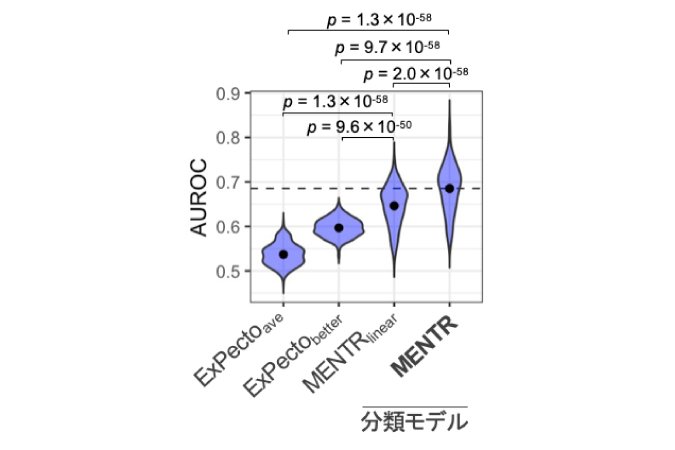
図1 エンハンサーRNA(eRNA)の予測精度の比較
横軸に示した手法で、FANTOM5の347の組織・細胞種の予測能(AUROC)を同じテストデータを用いて比較した。ドットはn=347の平均値、p値はWilcoxonの符号順位検定(両側)。従来法(ExPecto)ではその設計上、ゲノムのプラス方向にもマイナス方向にも転写されるeRNAを予測できない(他の従来法も同様)。そこでExPectoaveでは両方向の予測値の平均を取り、ExPectobetterでは恣意的に正解データに発現が近い方を選択したが、いずれもMENTRの方が高い予測精度を示した。MENTRlinearは、MENTRの勾配ブースティング決定木の代わりに単純な正則化線形モデルを用いた平均を取ったものであり、エピジェネティックな状態の組合せ効果(非線形効果)は学習されない。
さらに、MENTRがCAGEトランスクリプトームデータにおいて偽陽性の予測(評価データでは発現していないが、発現ありと予測)をした転写物について、検出感度の高いNET-CAGE[9]による5種類の同一の細胞株の計測データを用いて再度精査すると、そのうち31~70%が実際には転写されていたことが明らかになりました。すなわち、実験的理由によりCAGEでは未検出だった転写物について、MENTRでは正しく予測できる場合があり、上述の評価用データを用いた予測精度は実際にはもう少し高いことが分かりました。
また、MENTRが非翻訳RNAの予測精度を高められた理由の解明にも挑み、MENTRが転写物から遠くの細胞種特異的なエピジェネティックな状態を重要視していること、エピジェネティックな状態が二つ以上組み合わさった際の特別な効果(非線形な効果)の考慮が非翻訳RNAの予測能を向上させたことなどを明らかにしました。この結果は、非翻訳RNAの発現制御に非常に遠くのエピジェネティックな状態が複雑に関与することがデータ駆動的に見いだされたことを意味しており、AIの予測結果の解釈が生物学的知見と見事に合致した事例になります。
続いて、MENTRを用いたインシリコ変異導入法による発現量変化(以下、変異効果量)の正確さについて、154人のリンパ芽球様細胞株の遺伝型とCAGEトランスクリプトーム結果から推定した多型の効果(ある多型が遺伝子発現量を増減させる効果)を正解データとして検証しました。まず、mRNAと非翻訳RNAの両方において、変異効果量が大きくなるにつれて、変異効果量が正解データにおける多型の効果と一致することを明らかにしました。これは、変異効果量の大きさが、予測の正しさを示していることを意味します。
また、実験で用いられた細胞株と同じ種類の細胞をインシリコ変異導入法で用いたとき、変異効果量の予測の正しさが最大化されました。これは、遺伝子発現制御機構が細胞種特異的であり、細胞特異的な発現パターンを学習する必要性を示すものです(図2)。変異効果量と正解データの一致率が80%に達するためには変異効果量が5%以上、一致率90%の精度に達するためには変異効果量が10%以上変化することが必要であることも分かり、今後の応用で必要となる精度に応じた閾値を決定できました。
さらに、集団のデータを用いないMENTRによる変異効果量は連鎖不平衡[13]に依存しないピンポイントな予測であり、同じく連鎖不平衡の影響を受けない実験的変異導入法の結果とよく一致していました。以上の検討により、MENTRを使って非翻訳RNAの発現に影響する多型をピンポイントかつ細胞種特異的に予測できることが示されました。
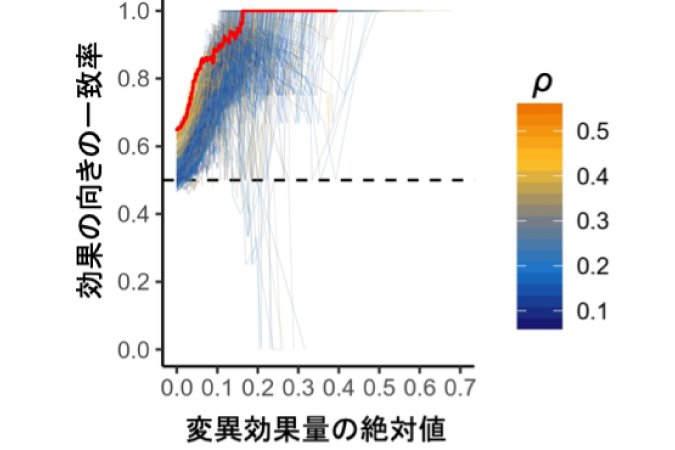
図2 変異効果量とその精度
リンパ芽球様細胞株におけるMENTRによるeRNAの変異効果量の閾値を変えたときに、既報(M Garieri et al. Nat Commun. 2017)のエンハンサーに関連する多型の効果をどの程度再現できるか一致率を評価した(赤線)。ヒートマップカラー(橙色〜青)はリンパ芽球様細胞株とFANTOM5の347の細胞種・組織間のeRNA発現パターンの相関を示し、橙色ほど発現パターンが似た細胞種であることを示している。全く異なるeRNA発現パターンを示す細胞種(青色)だと、既報との一致率が著しく低下した。つまり、インシリコ変異導入法で正確な変異効果を予測するためには、細胞種特異的な発現パターンの学習が必要不可欠である。
次に、MENTRを活用して、非翻訳RNAがヒトの複雑形質にどう寄与しているのかについて検証を進めました。これまで世界中で実施されたさまざまな複雑形質に対するGWAS結果を収集し、のべ41,223個の多型をMENTRのインシリコ変異導入法で解析したところ、17,306個(42%)の多型が少なくとも一つの転写物(mRNAを含む)の発現量の調節に関わることが分かりました(80%精度の閾値で選択)。一般集団が持つ多型全体では、わずか数%しかこの閾値に達する多型が存在しないことから、この発見確率は著しく高いものでした。この結果は、過去のGWAS結果の中に転写物の発現を変える変異が集積していることを意味し、従来のよく知られた知見と一致します。
また、これらの多型が影響する転写物には、lncRNAが3,548個、eRNAが7,775個(計11,323個)含まれていました。これらの非翻訳RNA(特にeRNA)の大多数は生物学的な機能が未解明であるため、GWAS結果を基に非翻訳RNAとさまざまな複雑形質とをリンクさせた本研究成果(カタログ)は、今後重要な研究リソースとなると考えられます。
実際にMENTRから新たにGWAS結果が解釈された実例を示します。ある多型(rs17293632)はクローン病[14]や喘息などと関連し(図3A)、SMAD3遺伝子のmRNAの発現量に影響することが報告されていました(図3B)。しかし、これらのゲノム解析では連鎖不平衡に依存して真の原因多型以外でも低いp値が観測されるため、原因多型を絞り込むのは困難でした。一方で、MENTRからは、クローン病に関係する大腸組織や喘息に関係する免疫関連細胞において、rs17293632のみが近傍のeRNAの発現を下げるとの予測(90%精度以上)がピンポイントで得られました(図3C)。実際、この多型がSMAD3遺伝子の転写活性に影響することは過去の実験にて報告されており、rs17293632がeRNAを介してSMAD3遺伝子のmRNAの転写活性を調節するメカニズムが裏付けられました。
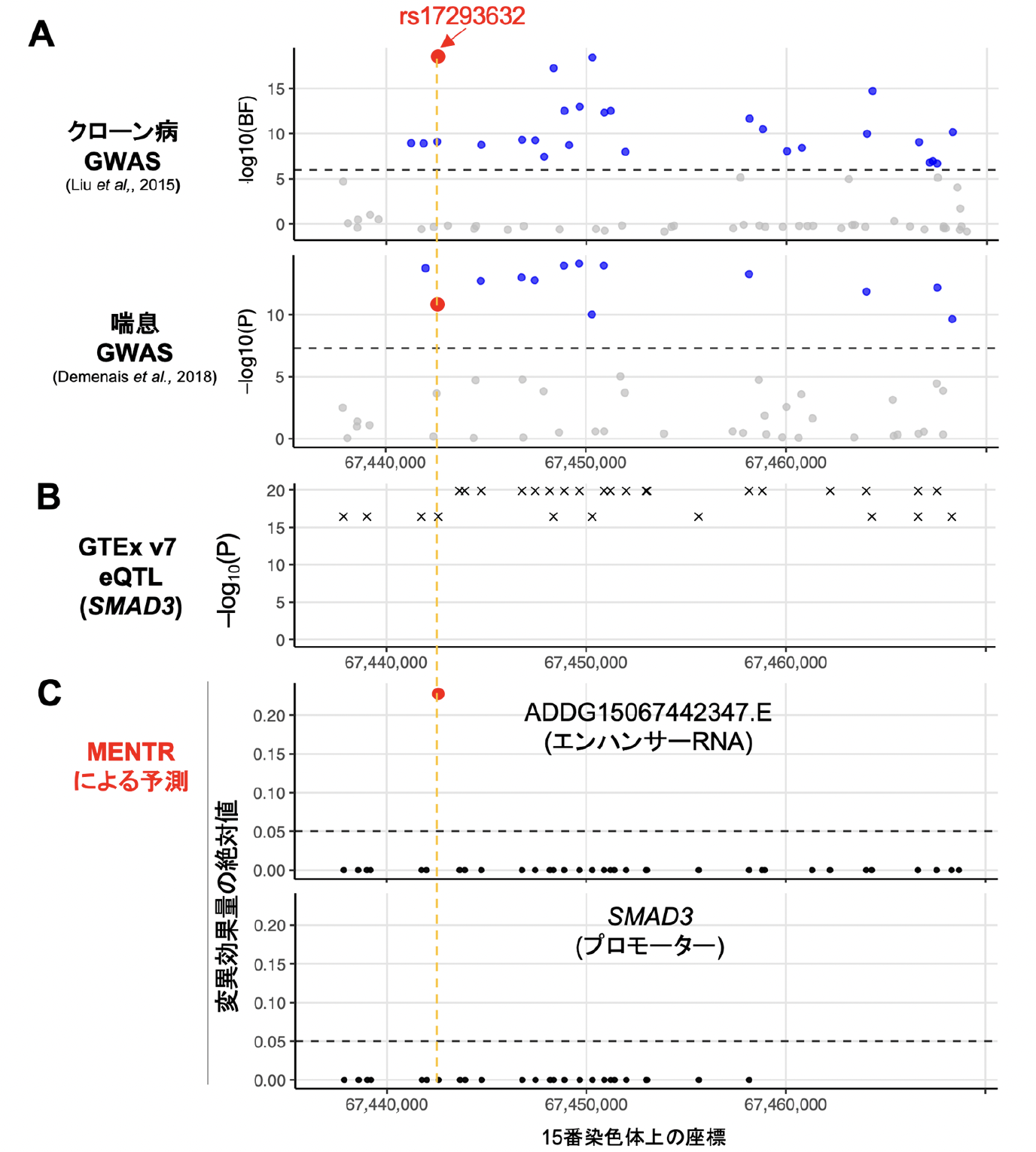
図3 MENTRのピンポイント予測の実例
(A) 連鎖不平衡に依存するGWAS結果(公開データから取得)。
(B) 連鎖不平衡に依存する遺伝子発現に関する量的形質座位(eQTL)解析結果(GTExデータベースから取得)。
(C) MENTRによるピンポイント予測(本研究成果)。
MENTRが最も必要とされるのは、多型の役割の根拠が存在しないような非常にまれな多型の解釈です。例えば、世界で1,000人に1人しか保有者がいないようなまれな多型であるrs570639864は踵(かかと)の骨密度を減少するとの報告がありますが、この多型が具体的にどのように骨密度低下につながるのかは明らかではありませんでした。興味深いことに、MENTRからは多くの細胞種で近傍のある1つのeRNAの発現量を下げると予測されました。従来の研究からは、このeRNAはWNT7B遺伝子のmRNAの転写活性と相関があること(このeRNAがWNT7B遺伝子の転写制御を行っていると考えられること)、そしてシグナル伝達物質として作用するWNT7Bタンパク質が骨形成を促進することがマウス研究から分かっていました。これらの知見を統合し、多型からeRNAを介して踵骨密度に影響するという生物学的解釈を得ることができました(図4A)。
また、まれな多型rs12722502は喘息の発症リスクを下げる効果が報告されていますが、MENTRからは特定の白血球におけるeRNAの発現を低下させることが予測されました。従来の研究から、このeRNAはIL2RA遺伝子のmRNAの転写活性と相関があることが知られており、興味深いことにIL2RAタンパク質を標的とするモノクローナル抗体(Daclizumab)[15]が肺機能と喘息を改善するとの報告があります(図4B)。以上のように、MENTRを活用することで、まれな多型がeRNAを介して複雑形質に関わるメカニズムが解釈可能になることが分かりました。
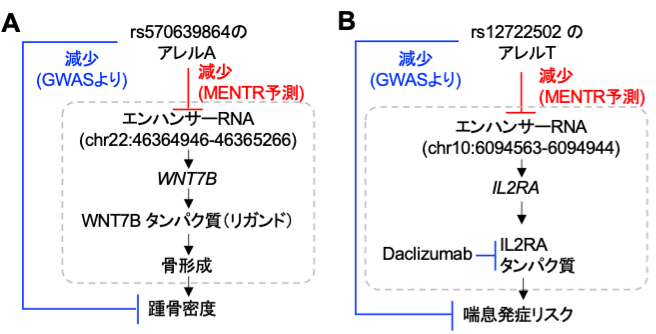
図4 複雑形質に関連するまれな多型のMENTRによる解釈例
赤: MENTRによる予測。青: GWASから得られる遺伝的関連。通常の矢印は+の効果の関係性(増加)、T型の矢印は-の効果の関係性(減少、阻害)を示す。
今後の期待
GWASから得られた多型と疾患との因果関連を示すデータが日進月歩で蓄積されていますが、GWAS関連領域の大多数を占める非翻訳領域上の多型が疾患発症リスクを増減する生物学的メカニズムを明らかにしたものは限定的です。DNA配列のみから細胞種特異的な非翻訳RNAの発現予測を行う新しいAI技術MENTRを用いると、多型による非翻訳RNAの発現量の増減を高精度に予測でき、創薬標的の探索に有用な生物学的解釈を行うことができます。現在、日本を含む世界各国で数万~数十万といった大規模なヒトの全ゲノムシーケンス解析[16]が進行中であり、今後もGWASから複雑形質に関連するまれな多型が非翻訳領域において多数明らかになると考えられます。そのため、創薬研究やバイオマーカー探索におけるMENTRの意義が一層高まるものと期待できます。本発表以外にも、MENTRは既に複数の研究で新知見を見いだすことに貢献しており注1-3)、今後のさらなる活用が期待できます。
同時に、「MENTRがなぜDNA配列のみから非翻訳RNAの発現を予測できるのか」は本研究の過程で生まれた新たな問いです。本研究では、転写開始点から遠方のエピジェネティックな状態が複雑に(非線形に)組み合わさって、非翻訳RNAの発現が決まることをデータ駆動的に見いだしました。今後、MENTRの学習済み機械学習モデルを精緻に分析することで、非翻訳RNAの発現メカニズムを細胞種特異的に解明できれば、分子細胞生物の新知見が創出されることも期待できます。
本研究で主に用いたデータはFANTOM5が2014年などに発表した研究成果ですが、創薬において有用な新知見を8年近く経過した今でも得ることができました。MENTRのようなAI技術をフル活用し、過去に蓄積されたデータを再解析することで、新知見が創出される研究が進展することが今後も期待できます。
MENTRは公開レポジトリGitHubにて公開しており注4)、本研究で構築したカタログ注5)とともに、世界中の研究者が誰でも利用することができます。
注1)Mishra et al., Stroke genetics informs drug discovery and risk prediction across ancestries. Nature (2022)
注2)2021年6月9日プレスリリース「アトピー性皮膚炎発症の新しい遺伝因子」
https://www.riken.jp/press/2021/20210609_2/index.html
注3)2021年8月19日プレスリリース「日本人の鼠径ヘルニア(脱腸)に関わる遺伝子座を同定」
https://www.riken.jp/press/2021/20210819_3/index.html
注4)MENTR (mutation effect prediction on ncRNA transcription)
https://github.com/koido/MENTR
注5)MENTR results viewer
https://doi.org/10.5281/zenodo.5638259
研究支援
本研究は、日本学術振興会(JSPS)科学研究費助成事業若手研究「多因子疾患のエピスタシス効果を同定するためのゲノム配列の深層学習戦略」(20K15773、研究代表者:小井土 大)、同基盤研究(A)「エンハンサーの遺伝的発現制御の解明による免疫疾患解析」(JP20H00462、研究代表者:寺尾 知可史)、日本リウマチ学会(JCR)次世代基礎研究推進プログラム研究助成「脊椎関節炎解析の基盤となる日本人の組織特異的遺伝子発現とエンハンサーマップの構築」、日本医療研究開発機構(AMED)難治疾患実用化研究事業「シングルセル統合ゲノミクス解析が解き明かす強皮症の病態基盤の開発」(JP21kk0305013、研究開発代表者:寺尾 知可史)、同ゲノム医療実現推進プラットフォーム・先端ゲノム研究開発事業「先天的/後天的構造多型に着目した免疫/精神疾患病態解明に関する研究開発」(JP21tm0424220、研究開発代表者:寺尾 知可史)、同革新的がん医療実用化研究事業「体細胞モザイクのがん発症および予後因子としての意義解明の開発」(JP21ck0106642、研究開発代表者:寺尾 知可史)による助成を受けて行われました。また、産総研のAI橋渡しクラウド(ABCI)を利用して得られた成果です。
論文情報
<タイトル>
Prediction of the cell-type-specific transcription of non-coding RNAs from genome sequences via machine learning
<著者名>
Masaru Koido, Chung-Chau Hon, Satoshi Koyama, Hideya Kawaji, Yasuhiro Murakawa, Kazuyoshi Ishigaki, Kaoru Ito, Jun Sese, Nicholas F. Parrish, Yoichiro Kamatani, Piero Carninci, Chikashi Terao
<雑誌>
Nature Biomedical Engineering
<DOI>
10.1038/s41551-022-00961-8
用語説明
[1] 非翻訳RNA、長鎖ノンコーディングRNA(lncRNA)
DNAから転写されるRNAのうち、メッセンジャーRNA(mRNA)はタンパク質に翻訳される。タンパク質に翻訳されるmRNAに対して、タンパク質に翻訳されないRNAの総称を非翻訳RNAという。非翻訳RNAは、ヒトの発生や恒常性の維持などにおいて、組織や細胞種特異的に作用する様々なタイプがあることが報告されている。本研究ではこのうち、遺伝子の発現を遠方から制御するエンハンサーが活性化した際にそのエンハンサー領域の両端から同時に転写されるエンハンサーRNA([7])と、200塩基以上の長鎖ノンコーディングRNA(lncRNA)を主な解析対象とした。lncRNA はlong non-coding RNAの略。
[2] AI
人工知能(artificial intelligence)の略。本研究では特に、データとその学習ルールを指定して、変数間の複雑な関係性・規則性を学習する技術である機械学習(Machine Learning)を指す。
[3] 多型
ゲノムのDNA配列が個人間で異なる箇所のうち、ある集団内で一定の頻度で存在するもの。代表的なものに一つの塩基が他の塩基に置き換わった一塩基多型(Single Nucleotide Polymorphism、SNP)がある。
[4] 配列モチーフ、転写因子
特定の機能的役割を持つDNA配列パターン。例えば、ゲノム上にある転写因子(特定のDNA配列に結合し、遺伝子発現を制御するタンパク質)が結合する箇所は数多く存在するが、その結合箇所のDNA配列パターンを比較すると同じような配列が頻出する。例えば、2019年のノーベル医学生理学賞で話題になった低酸素誘導因子はACGTGやGCGTGなどの5文字のモチーフがある領域に結合することが知られている。そのような頻出パターンの最小単位として配列モチーフが用いられてきたが、本研究ではヒトが発見できないような長くて複雑な配列パターンも含めてAI技術で探索を試みた。
[5] FANTOMコンソーシアム
理研が主催する国際研究コンソーシアム(https://fantom.gsc.riken.jp/jp/)。本研究では、第5期(FANTOM5)で公開されたヒトのさまざまな細胞・組織で計測したCAGEトランスクリプトームデータを再解析し、用いた。
[6] ゲノムワイド関連解析(GWAS)
多因子疾患や身体測定値などのさまざまな複雑形質に対して、遺伝多型との関連を明らかにするために行われるゲノム解析手法。GWASはGenome-Wide Association Studyの略。
[7] エンハンサーRNA(eRNA)
エンハンサーは、遺伝子発現を制御する機能部位のうち、転写開始点から遠方に存在し、転写効率を高めるのに重要な領域。同じmRNAに対しても、細胞種が異なれば別の種類のエンハンサーが機能している。特に、活性化したエンハンサーの両端からは非翻訳RNAが転写されることが知られており、これをエンハンサーRNA(eRNA)と呼ぶ。FANTOMコンソーシアム第5期(FANTOM5)によって65,000個のeRNAが同定され、その周辺に多因子疾患の関連多型が数多く存在することが報告されている。
[8] ディープラーニング
機械学習方法の一種で深層学習とも呼ばれる。ニューラルネットワークと呼ばれる生物の神経回路を模倣した機械学習モデルを何層も繰り返すことで、さまざまな特徴量を作り出しつつ、学習がうまくいくようなさまざまな工夫を重ねることで、高精度な予測を実現する。特に本研究では、画像認識で用いられてきた深層畳み込みニューラルネットワークを用いている。DNA配列の場合、局所的な情報(例えば連続する3塩基)の重み付き和を計算して、新たな特徴量を作り出す"畳み込み"を、DNA配列全体にわたって実施する。この畳み込みを含む処理を何度も繰り返したディープラーニングモデルを深層畳み込みニューラルネットワークと呼ぶ。
[9] CAGEトランスクリプトームデータ、NET-CAGE
CAGE(Cap Analysis Gene Expression)は理研が開発した転写物の5'末端の塩基配列を決定する手法。トランスクリプトームは対象とした細胞・臓器などに存在している全ての転写物の集合・総体のこと。つまり、CAGEトランスクリプトームデータは、CAGEで測定した転写物全体のデータを指す。NET-CAGE(Native Elongating Transcript-CAGE)は新しく合成されている最中の新生RNAを迅速かつ高純度で精製する生化学手法を用いたCAGE 。
[10] エピジェネティックな状態量
DNAのメチル化やヒストン修飾などの後天的に変化する化学的修飾が遺伝子発現制御に関わると考えられており、その化学的修飾の活性化(エピジェネティックな状態)の程度を指す。臓器・細胞種ごとに特徴的なパターンを示すことが広く知られている。本研究では、深層畳み込みニューラルネットワークが出力する細胞別のヒストンメチル化や転写因子の結合など2,002種の状態量を用いた。
[11] 勾配ブースティング決定木
機械学習法の一つで、ディープラーニングと並んでデータ分析コンペティションでよく使用される手法。ベースとなるのは決定木と呼ばれるシンプルな機械学習方法であり、データの非線形な特徴も加味した予測が可能である。
[12] ROC曲線下面積
ROC(Receiver Operating Characteristic)曲線は、ある分類を行うとき、予測指標の閾値を変えたときに感度と特異度が変わっていく関係性を図示したもの。x軸に1-特異度、y軸に感度をプロットする。予測精度が高いほど曲線は左上に位置するため、その曲線下の面積が予測精度の指標として用いられる。予測能が高いほど、ROC曲線下面積は1に近づき、ランダムな予測指標であればROC曲線下面積は0.5となる。
[13] 連鎖不平衡
ある集団において、二つの多型の遺伝型に着目したとき、両者がランダムな関係でないこと。近傍の多型で連鎖不平衡はしばしば観察されるため、図3のAやBのように原因多型以外でも関連(低いp値)が得られる。MENTRでは集団を用いた解析ではないため、連鎖不平衡に依存しない多型の影響をピンポイントに予測できる。
[14] クローン病
炎症性腸疾患の難病の一つ。大腸や小腸に炎症または潰瘍が起こりやすい。
[15] モノクローナル抗体(Daclizumab)
活性化T細胞で発現するヒトのIL2受容体αサブユニット(別名 CD25)に対する抗体製剤。
[16] 全ゲノムシーケンス解析
次世代シーケンサーを用いて、全ゲノムDNAの配列を解読すること。