森下 真一
(もりした しんいち/教授/生命科学研究系)
メディカル情報生命専攻/バイオ情報科学大講座/バイオデータベース分野

略歴
1983年3月 東京大学理学部情報科学科卒業.
1985年3月 東京大学大学院理学系研究科情報科学専攻修士課程修了.
1985年4月 日本IBM(株)入社.
1990年2月 理学博士(東京大学).
1990-2年スタンフォード大学計算機科学科およびIBM アルマデン研究所にて在外研究.
1997年9月 東京大学医科学研究所ゲノム知識発見システム(日立)寄付研究部門客員助教授.
1999年9月より新領域創成科学研究科・複雑理工学専攻・助教授. 理学部情報科学科・助教授(兼担).
2003年4月より現職.
1983 BS, Department of Information Science, Faculty of Science, University of Tokyo, Japan
1985 MS, Department of Information Science, Graduate School of Science, University of Tokyo, Japan
1985-97 Researcher, IBM Japan
1990 PhD, Department of Information Science, Graduate School of Science, University of Tokyo, Japan
1990-92 Visiting Researcher, Department of Computer Science, Stanford University / IBM Almaden Research Center
1997-2000 Visiting Associate Professor, Institute of Medical Science
1999-2003 Associate Professor, Department of Complexity Science and Engineering, Graduate School of Frontier Sciences, University of Tokyo / Adjunct Associate Professor, Department of Information Science, Faculty of Science, University of Tokyo
2003- Professor, Department of Computational Biology, Graduate School of Frontier Sciences, University of Tokyo
教育活動
大学院:情報生命科学基礎 I, 情報生命科学演習
理学部:生物情報学基礎論 I, 生物情報ソフトウエア論, 生物データマイニング論, 生命情報表現論, 生物情報科学情報基礎実験
研究活動
1) ゲノム解読と進化
DNA(ゲノム)には幅広いスケールの変化が起こることが知られています. 数億年にわたっておこる染色体レベル大規模な再編成, 数万年間の塩基レベルの小さな変化, さらに発生段階のDNA修飾等があり, これらが遺伝子の機能を豊かにし多様な表現型の生物を生んだと考えられています. 「DNAにはどのような変化が起きているのか?」この疑問を解くため, 過去10年間, 膨大なDNAデータと変異体の顕微鏡画像データを収集し, 情報分析するアルゴリズムを研究してきています.
哺乳類クラスのDNAを解読することは, 数千万ピースのジグソーパズルを解くようなものです. しかも元の絵も教えてもらえない難題です. 2002年ごろに研究を開始し, 2005年ごろには並列計算機を活かした実用的ソフトウェアが出来上がりました(文献1). 哺乳類のDNA解読はアメリカ主導でしたが, 我々も独力でメダカとカイコ等の大型DNAを解読できるようになりました (文献2). 副産物として, 過去 6~10億年にわたって脊椎動物や昆虫のDNAが進化してきた様子を描出することができました(図1). 特に「脊椎動物初期にDNA全体が2度重複し遺伝子セットを豊かにした」という1970年にOhnoが推測した現象を証明できました (文献3).
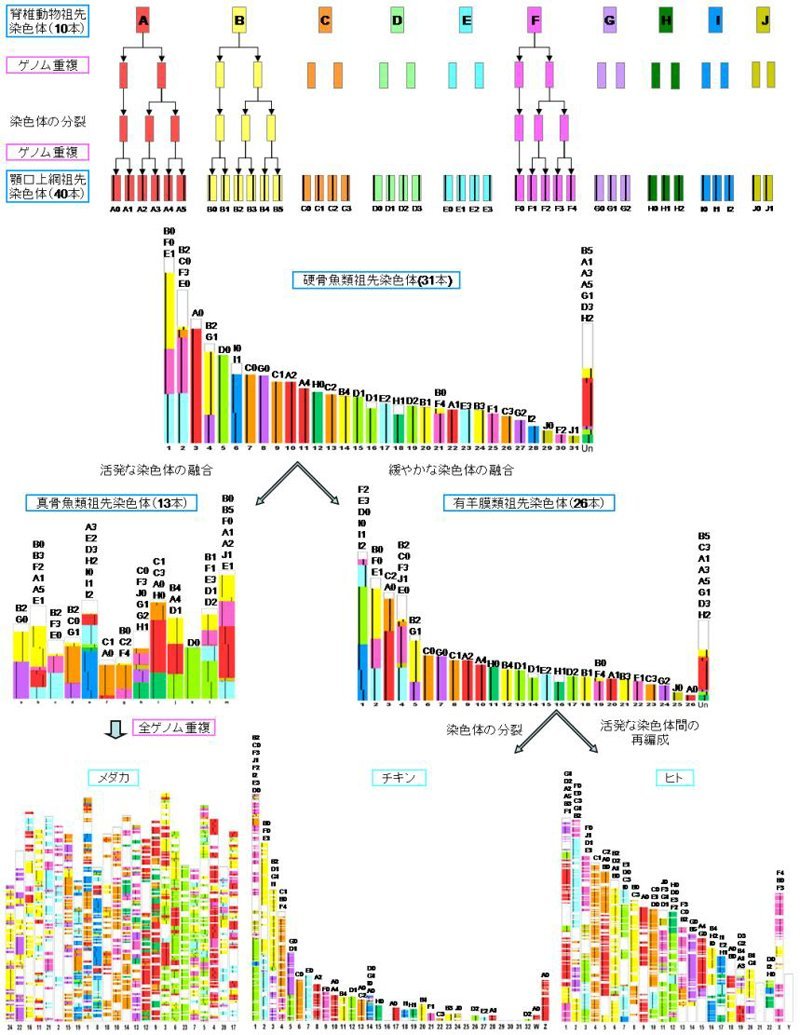
脊椎動物ゲノム進化の全体像の一部
2) ゲノム情報ビッグバンから読み解く生命圏
一方, 数百万年から数万年のスケールでは, 置換, 挿入, 削除等の塩基レベルの変化も異なる表現型(例:病気, 薬の副作用等)に関与している可能性があります. そこで長大なDNAを比較して違いを調べることが重要になるわけですが, 最近になってようやく可能になってきました. 理由は, 2007年頃より普及してきた超高速DNA解読装置にあります. 2009年8月現在 10-20億塩基/日の解読能力を持ち, 2002年からの7年間で約1000倍も性能が伸びました(文献4). この革命的進歩は「ゲノム情報ビッグバン」と呼ばれています. この進歩に対応するため, 情報生命科学専攻が中心となって提案したグローバルCOEプログラム「ゲノム情報ビッグバンから読み解く生命圏」が 2009年度に採択されています.
3) 超並列計算とゲノム情報ビッグバン
ナノバイオ技術の進展は著しく, 2011年ごろには, 塩基生産能力がさらに現在の1000倍近く(2兆塩基/日)の装置が普及しても不思議でありません. ムーアの法則(1.5年間でCPU性能が約2倍)を遙かに凌駕するスピードなので, 多数の計算機を並べて実行する高速化が要となります. 私たちも常時1000 CPUコア以上の計算機資源を使っており, 今後も並列化アルゴリズムの研究がDNA研究のカギとなるでしょう.

ゲノム情報ビッグバンのイメージ
4) ゲノムの高次構造の理解に向けて
超高速DNA解読装置の応用範囲はきわめて広く, DNAの比較はむろんのこと, 以前は全く手が出せなかった問題への応用が可能になってきています. 従来はゲノムの1次元配列構造の理解が精一杯でしたが, より高次な構造の理解に手が届きつつあります. たとえば, DNAのどの部分がヌクレオソームに巻きついているかというクロマチン構造の描出, DNAのメチル化の状態と遺伝子の転写メカニズムの関連, などが調べやすくなりました. 私たちが最近報告した面白い現象は, 転写開始点下流において, 遺伝的多様性には周期性があり, クロマチン構造と相関するという結果です(文献5). クロマチン構造が進化に影響する新しい視点が歓迎されました(文献6).
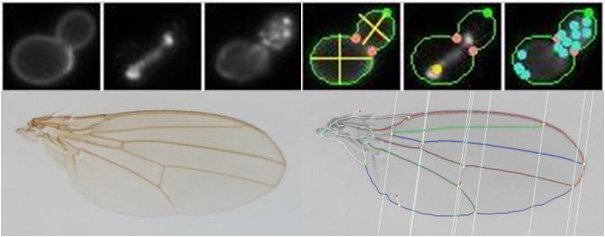
形態異常を検出するイメージ処理ソフトウエアの解析結果
5) 表現型幾何学の展開, フェノーム画像分析ソフトウエア
遺伝子の破壊や強制発現が形態にもたらす変化を緻密に定量化すれば, 統計学的な根拠から変異体の特徴を記述でき, 遺伝子機能の理解に役立つと私たちは考えています. そこで出芽酵母のすべての非必須遺伝子の破壊株の顕微鏡写真から, 細胞壁・核・アクチンの微小な形態異常をも検出するイメージ処理ソフトウエアを研究開発し, いくつかの遺伝子の機能を推定しました(文献7). またショウジョウバエにおいて遺伝子を強制発現させたとき翅脈にどのような形態変化が起こるかについても同様の分析を行っています.
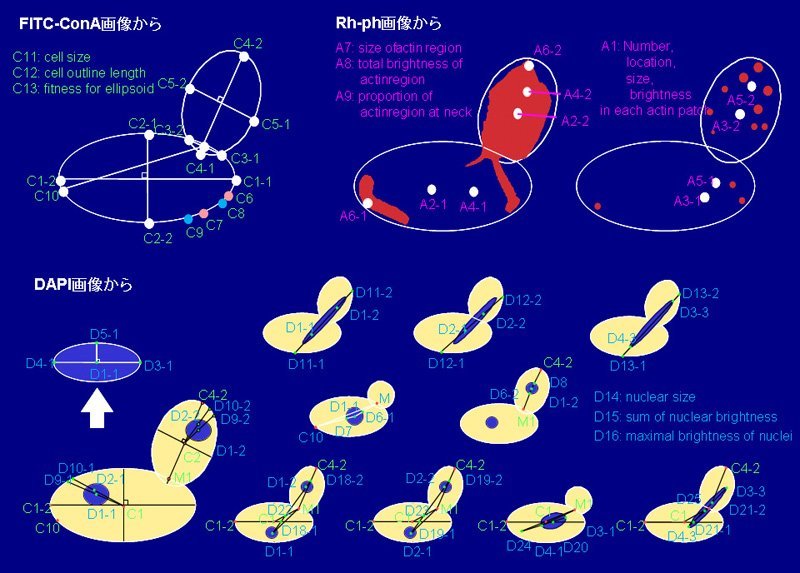
表現型の幾何学的パラメータ
6) データベースおよびデータマイニング研究
1990年代からデータベースおよびデータマイニングの研究を行ってきており, 関心のある大学院生をこの方面の研究で指導しています(文献8,9など). どちらの分野も生命科学研究との関連が密接です. 特にゲノム情報ビッグバン時代の大量データ分析にはデータベースおよびデータマイニングのアルゴリズムとソフトウエアが幅広く応用されています.
文献
1) Kasahara, M. and Morishita, S.: Large-scale genome sequence processing, Imperial College Press, 248p, 2006.
2) Kasahara, M., et al., Nature, 447:714-719, 2007.
3) Nakatani, Y., et al., Genome Research, 17(9):1254-1265, 2007.
4) 生命研究への応用と開発が進む バイオデータベースとソフトウェア最前線(森下・阿久津編)『実験医学』増刊号2008年4月
5) Sasaki, S. et al. Science, 323(5912):401-404, 2009.
6) Semple et al. Science, 323(5912):347-348, 2009.
7) Ohya, Y., et al., PNAS, 102(52):19015-20, 2005.
8) Morishita, S. et al., ACM PODS, 2000.
9) Saito, T. et al., ACM SIGMOD, 2008.
その他
海外では ACM-SIGMOD, VLDB, WWW, ACM-SIGKDD, ACM-CIKM などの国際会議のプログラム委員, ACM SIGKDD カリキュラム委員等を十数年間務め, 学会活動をお手伝いしています. 国内では,日本ソフトウエア科学会評議員, バイオインフォマティクス学会評議員. 情報処理学会論文誌編集委員会主査(1998-9年), 日本ソフトウエア科学会理事(1998年-2002年) などです.
将来計画
生物学, 医科学におけるオーミクスデータ(ゲノム,トランスクリプトーム,プロテオーム,フェノーム)を解析するための基礎理論とバイオインフォマティクス・ソフトウエアの研究開発を推進してゆく計画です.
教員からのメッセージ
基本的なバイオインフォマティクス・ソフトやアルゴリズムを, できるだけ自分たちでつくり広める研究を応援しています.